

Betacoronavirus-specific alternate splicing
- PMID: 35074468
- PMCID: PMC8782732
- DOI: 10.1016/j.ygeno.2022.110270
Betacoronavirus-specific alternate splicing
Abstract
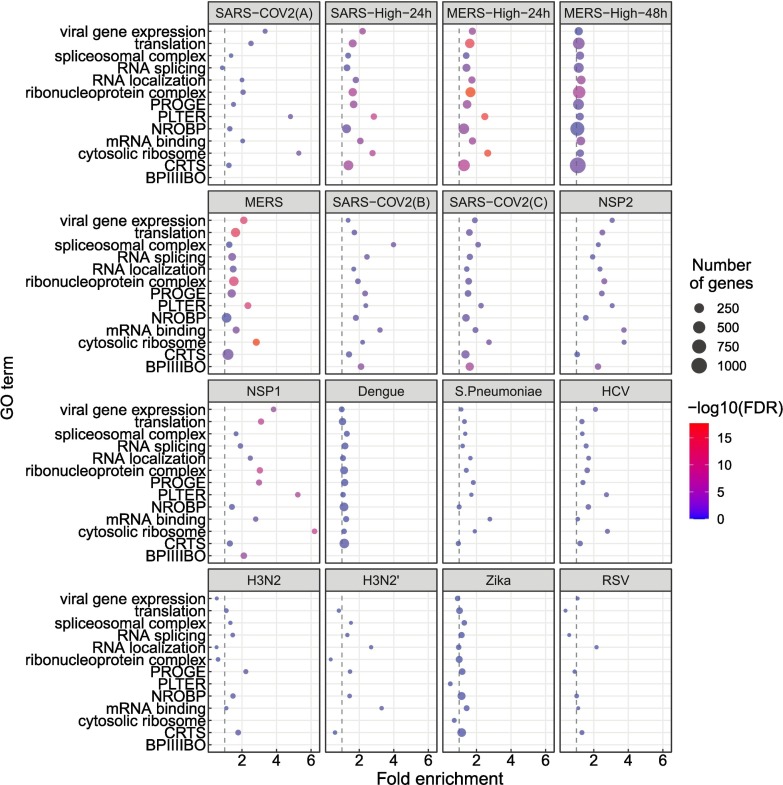
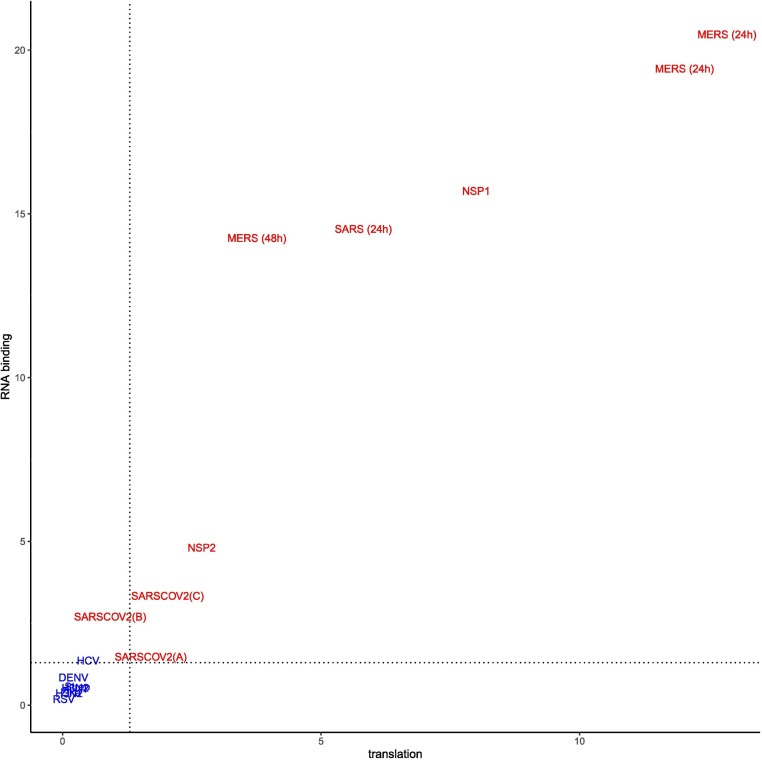
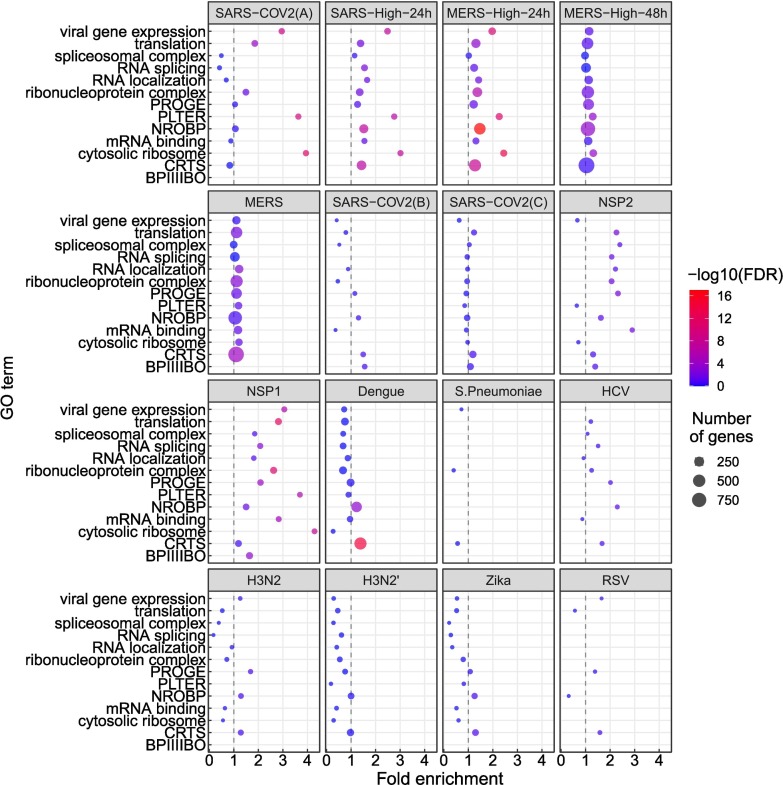
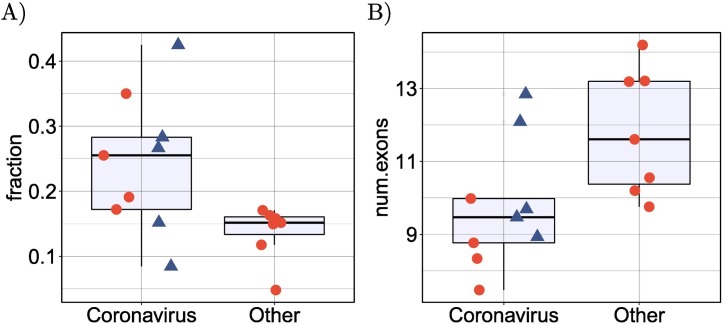
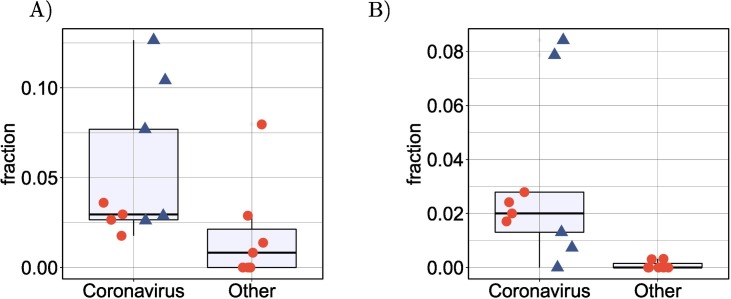
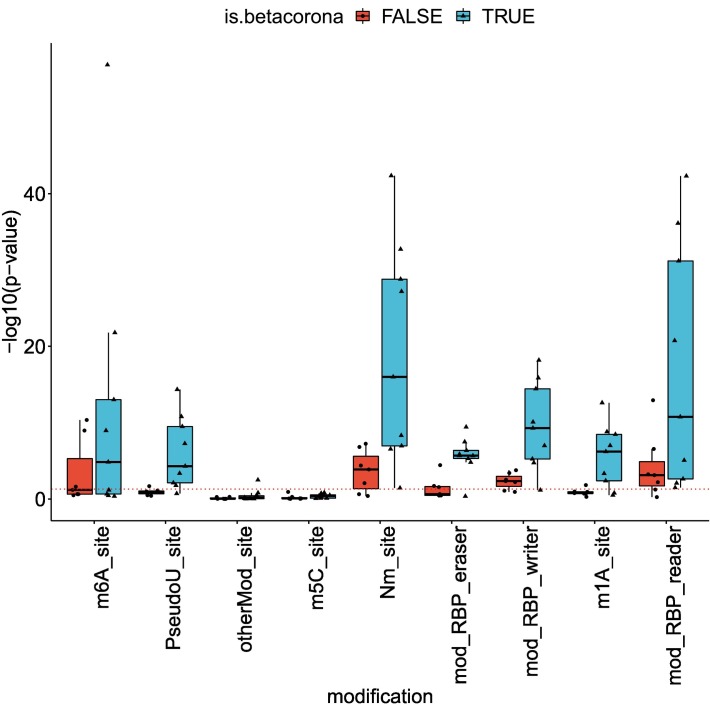
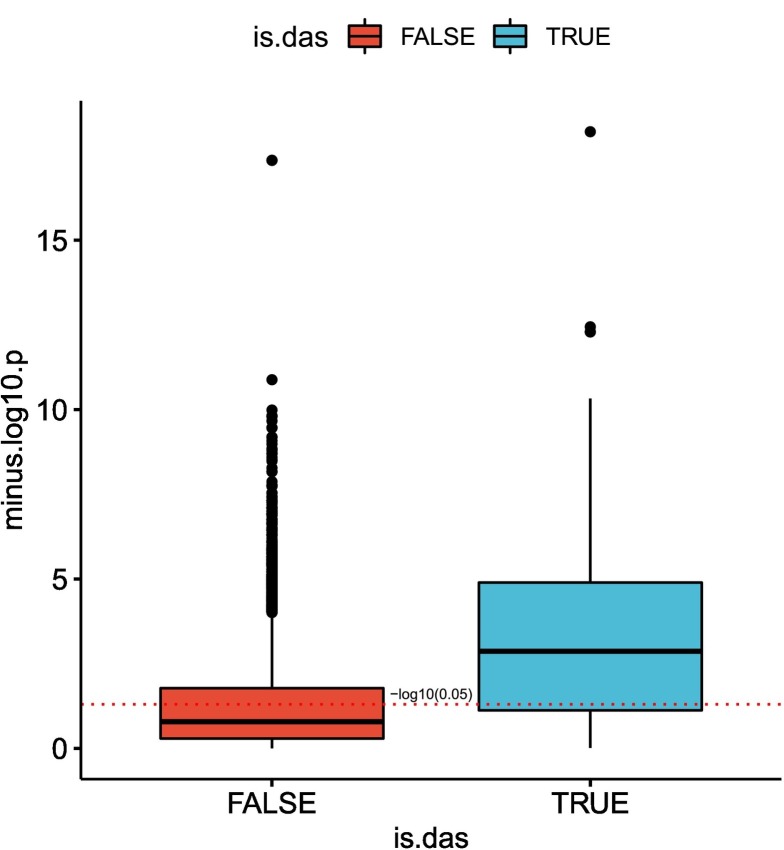
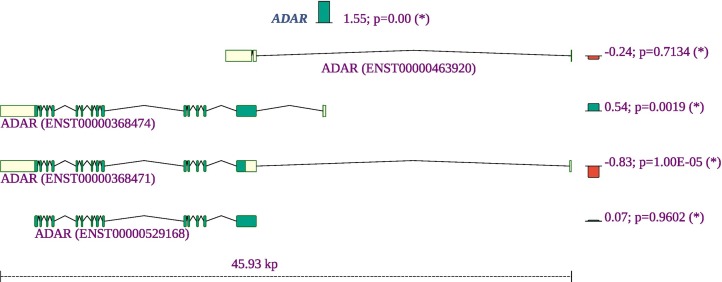
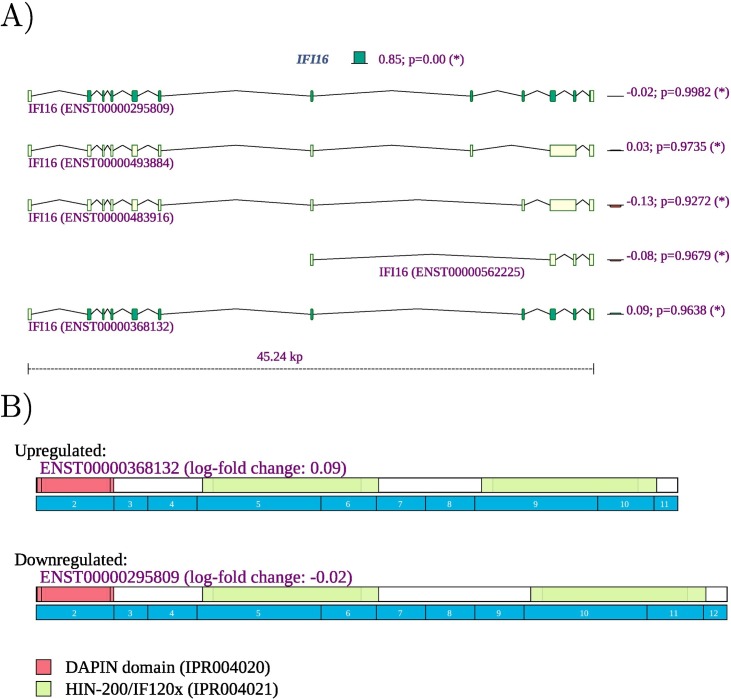
Viruses can subvert a number of cellular processes including splicing in order to block innate antiviral responses, and many viruses interact with cellular splicing machinery. SARS-CoV-2 infection was shown to suppress global mRNA splicing, and at least 10 SARS-CoV-2 proteins bind specifically to one or more human RNAs. Here, we investigate 17 published experimental and clinical datasets related to SARS-CoV-2 infection, datasets from the betacoronaviruses SARS-CoV and MERS, as well as Streptococcus pneumonia, HCV, Zika virus, Dengue virus, influenza H3N2, and RSV. We show that genes showing differential alternative splicing in SARS-CoV-2 have a similar functional profile to those of SARS-CoV and MERS and affect a diverse set of genes and biological functions, including many closely related to virus biology. Additionally, the differentially spliced transcripts of cells infected by coronaviruses were more likely to undergo intron-retention, contain a pseudouridine modification, and have a smaller number of exons as compared with differentially spliced transcripts in the control groups. Viral load in clinical COVID-19 samples was correlated with isoform distribution of differentially spliced genes. A significantly higher number of ribosomal genes are affected by differential alternative splicing and gene expression in betacoronavirus samples, and the betacoronavirus differentially spliced genes are depleted for binding sites of RNA-binding proteins. Our results demonstrate characteristic patterns of differential splicing in cells infected by SARS-CoV-2, SARS-CoV, and MERS. The alternative splicing changes observed in betacoronaviruses infection potentially modify a broad range of cellular functions, via changes in the functions of the products of a diverse set of genes involved in different biological processes.
Keywords: Alternative splicing; Betacoronavirus; COVID-19; Gene regulation; SARS-CoV-2.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures

Update of
-
Betacoronavirus-specific alternate splicing.bioRxiv [Preprint]. 2021 Jul 2:2021.07.02.450920. doi: 10.1101/2021.07.02.450920. bioRxiv. 2021. Update in: Genomics. 2022 Mar;114(2):110270. doi: 10.1016/j.ygeno.2022.110270. PMID: 34230929 Free PMC article. Updated. Preprint.
References
-
- Puelles V.G., Lütgehetmann M., Lindenmeyer M.T., Sperhake J.P., Wong M.N., Allweiss L., Chilla S., Heinemann A., Wanner N., Liu S., Braun F., Lu S., Pfefferle S., Schröder A.S., Edler C., Gross O., Glatzel M., Wichmann D., Wiech T., Kluge S., Pueschel K., Aepfelbacher M., Huber T.B. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 2020;383(6):590–592. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
