

Disruption of MeCP2-TCF20 complex underlies distinct neurodevelopmental disorders
- PMID: 35074918
- PMCID: PMC8794850
- DOI: 10.1073/pnas.2119078119
Disruption of MeCP2-TCF20 complex underlies distinct neurodevelopmental disorders
Abstract
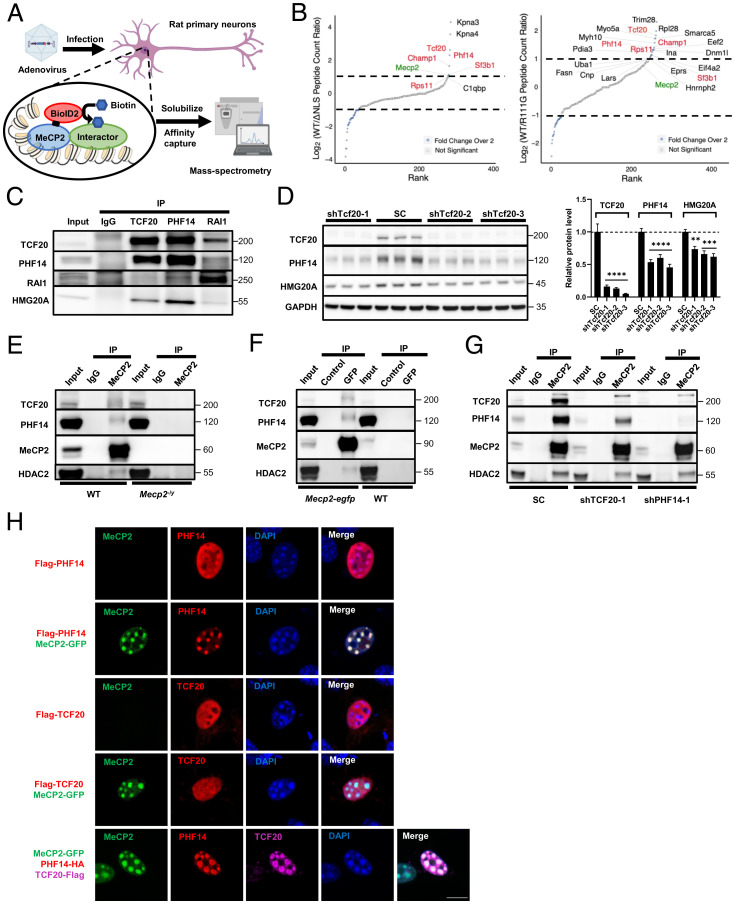
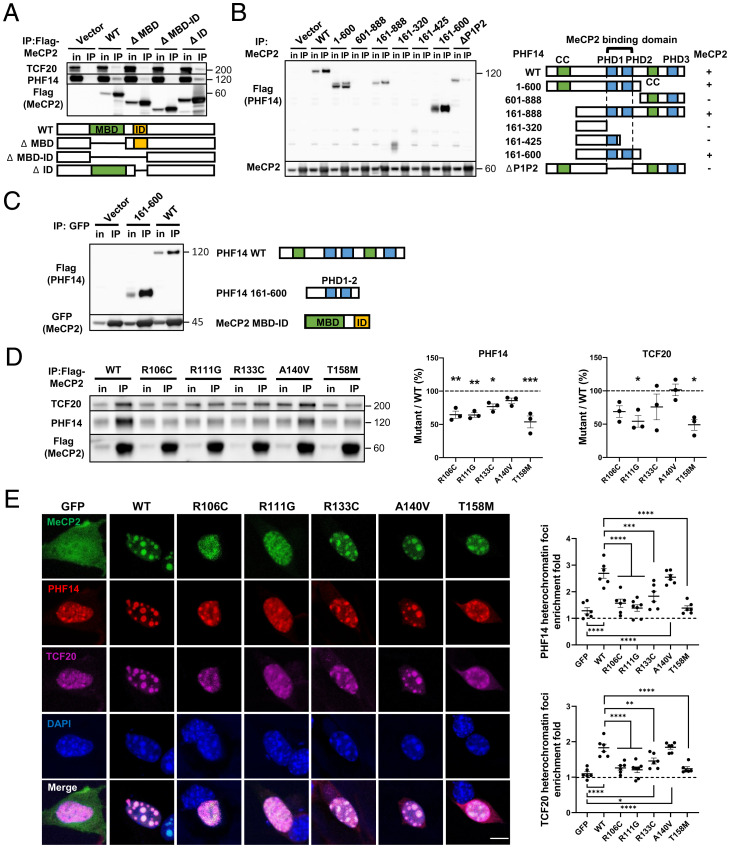
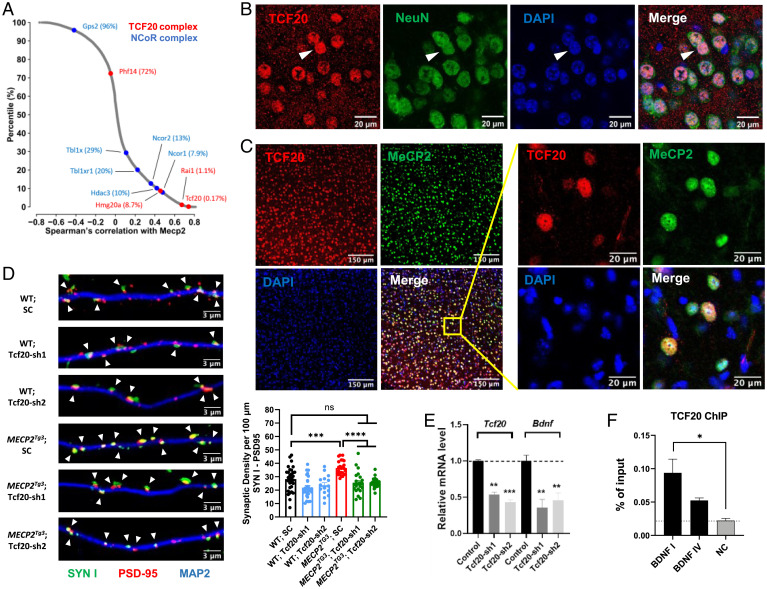
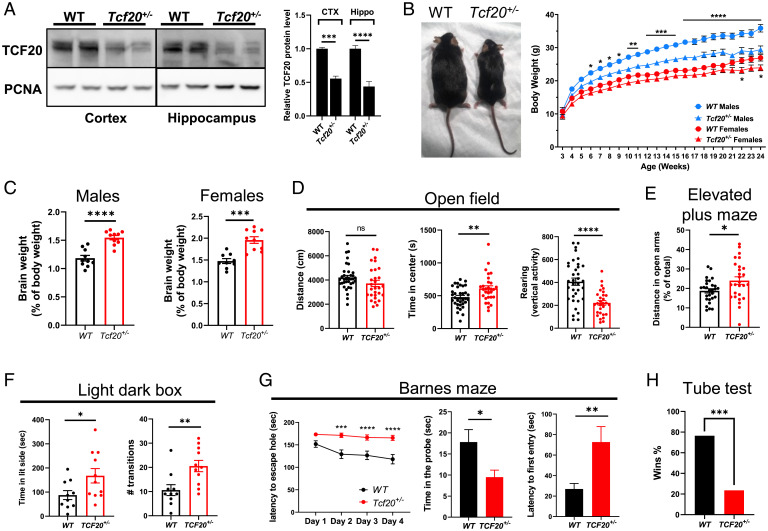
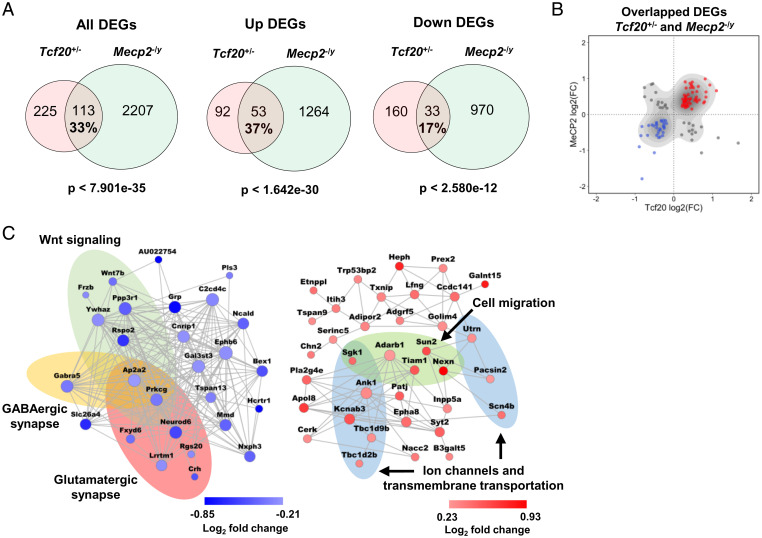
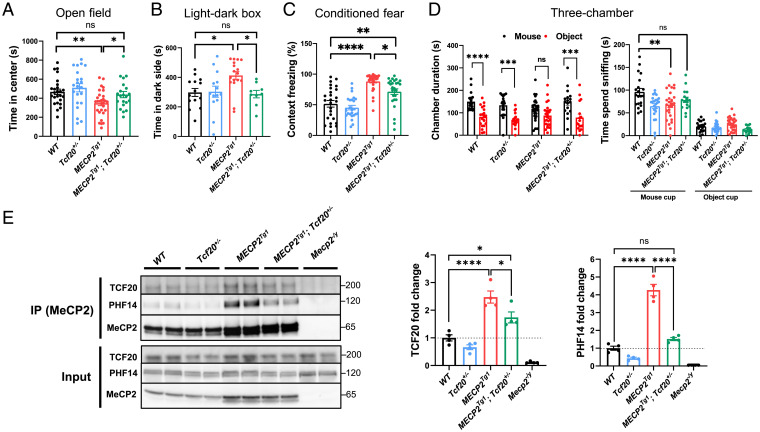
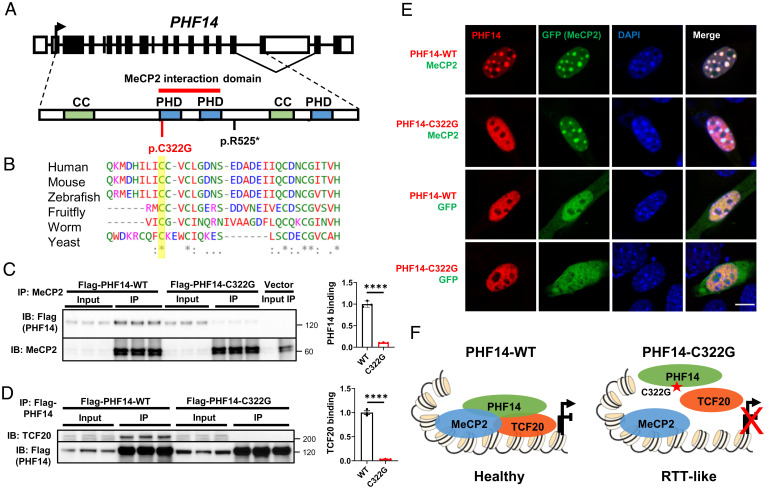
MeCP2 is associated with Rett syndrome (RTT), MECP2 duplication syndrome, and a number of conditions with isolated features of these diseases, including autism, intellectual disability, and motor dysfunction. MeCP2 is known to broadly bind methylated DNA, but the precise molecular mechanism driving disease pathogenesis remains to be determined. Using proximity-dependent biotinylation (BioID), we identified a transcription factor 20 (TCF20) complex that interacts with MeCP2 at the chromatin interface. Importantly, RTT-causing mutations in MECP2 disrupt this interaction. TCF20 and MeCP2 are highly coexpressed in neurons and coregulate the expression of key neuronal genes. Reducing Tcf20 partially rescued the behavioral deficits caused by MECP2 overexpression, demonstrating a functional relationship between MeCP2 and TCF20 in MECP2 duplication syndrome pathogenesis. We identified a patient exhibiting RTT-like neurological features with a missense mutation in the PHF14 subunit of the TCF20 complex that abolishes the MeCP2-PHF14-TCF20 interaction. Our data demonstrate the critical role of the MeCP2-TCF20 complex for brain function.
Keywords: BioID; MeCP2; Rett syndrome; TCF20 complex; neurodevelopmental disorders.
Copyright © 2022 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Amir R. E., et al. , Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188 (1999). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
