

Mechanical disruption of E-cadherin complexes with epidermal growth factor receptor actuates growth factor-dependent signaling
- PMID: 35074920
- PMCID: PMC8794882
- DOI: 10.1073/pnas.2100679119
Mechanical disruption of E-cadherin complexes with epidermal growth factor receptor actuates growth factor-dependent signaling
Abstract
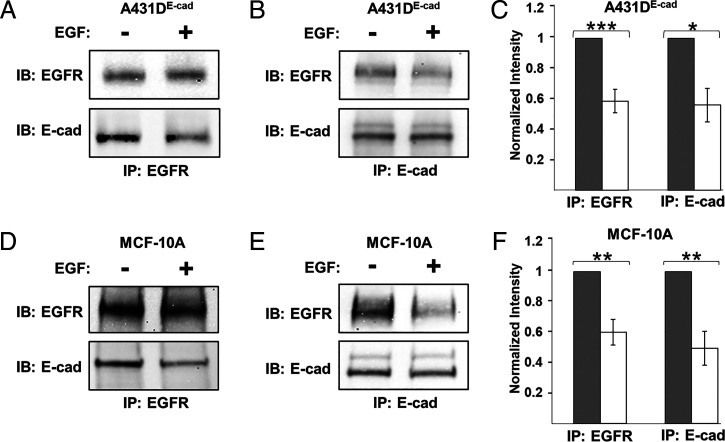
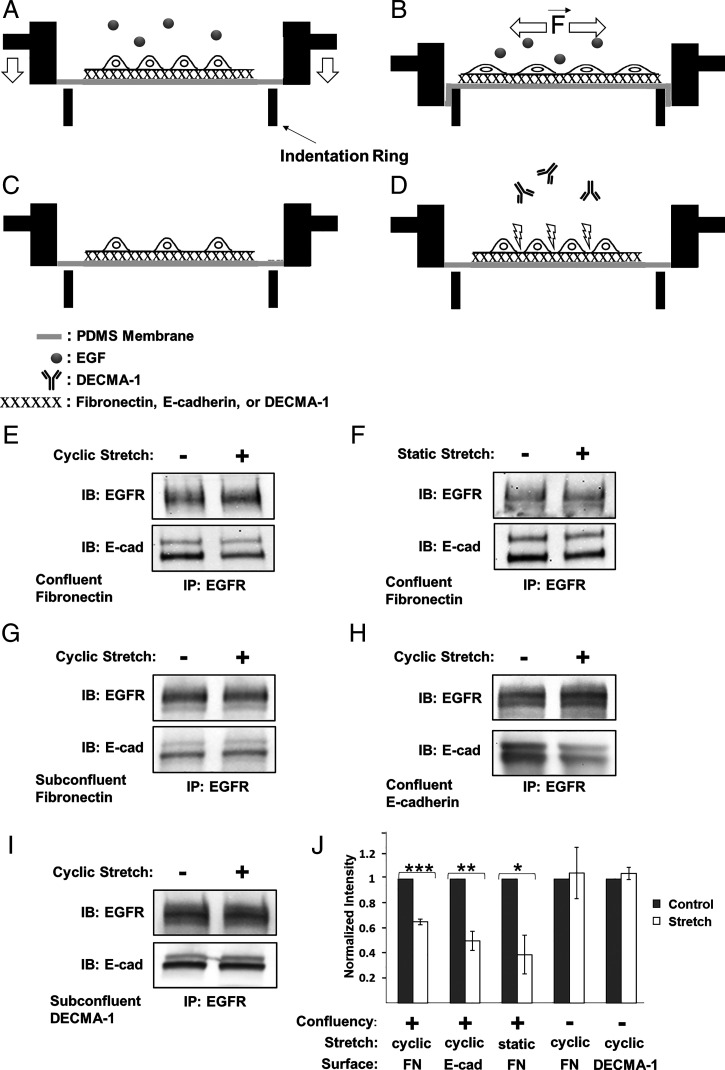
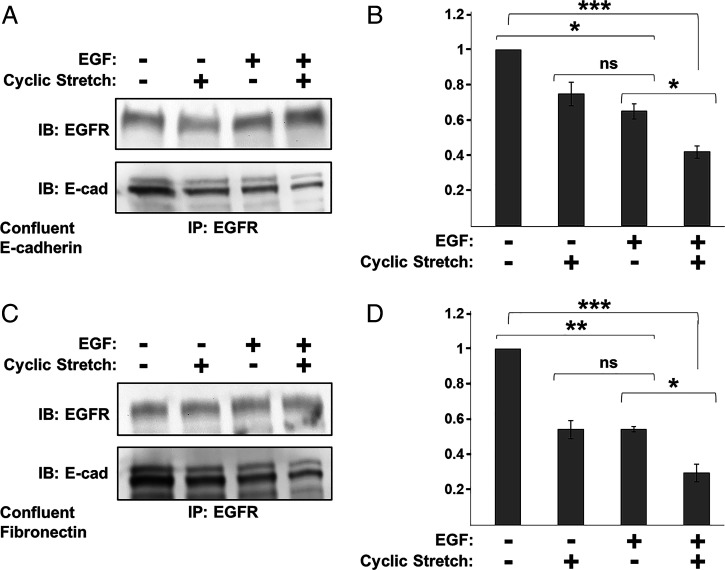
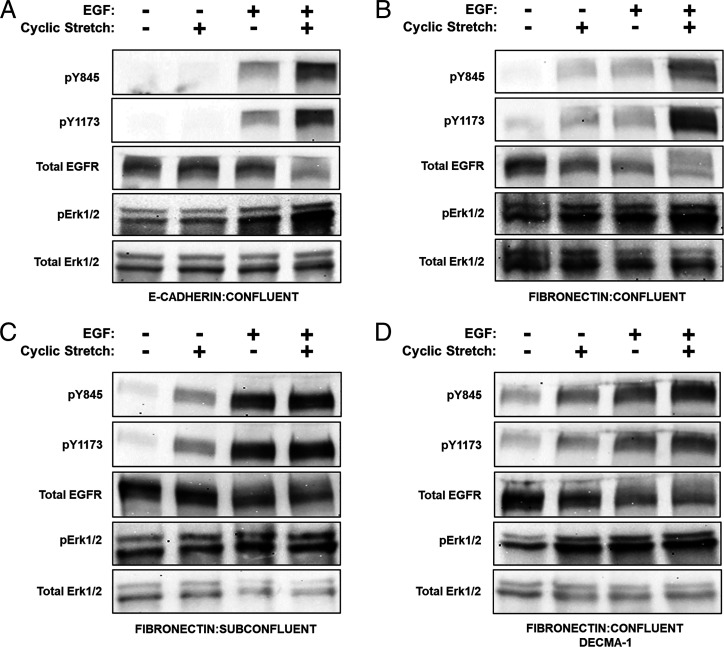
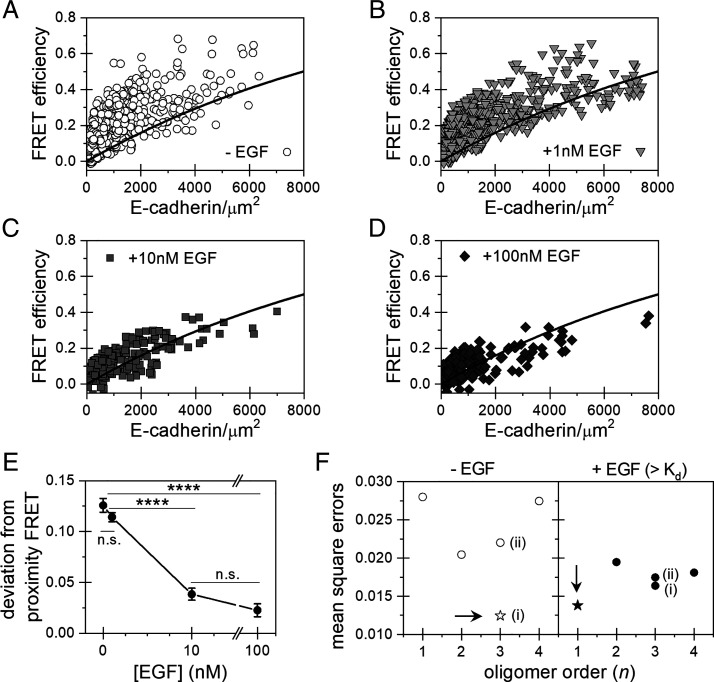
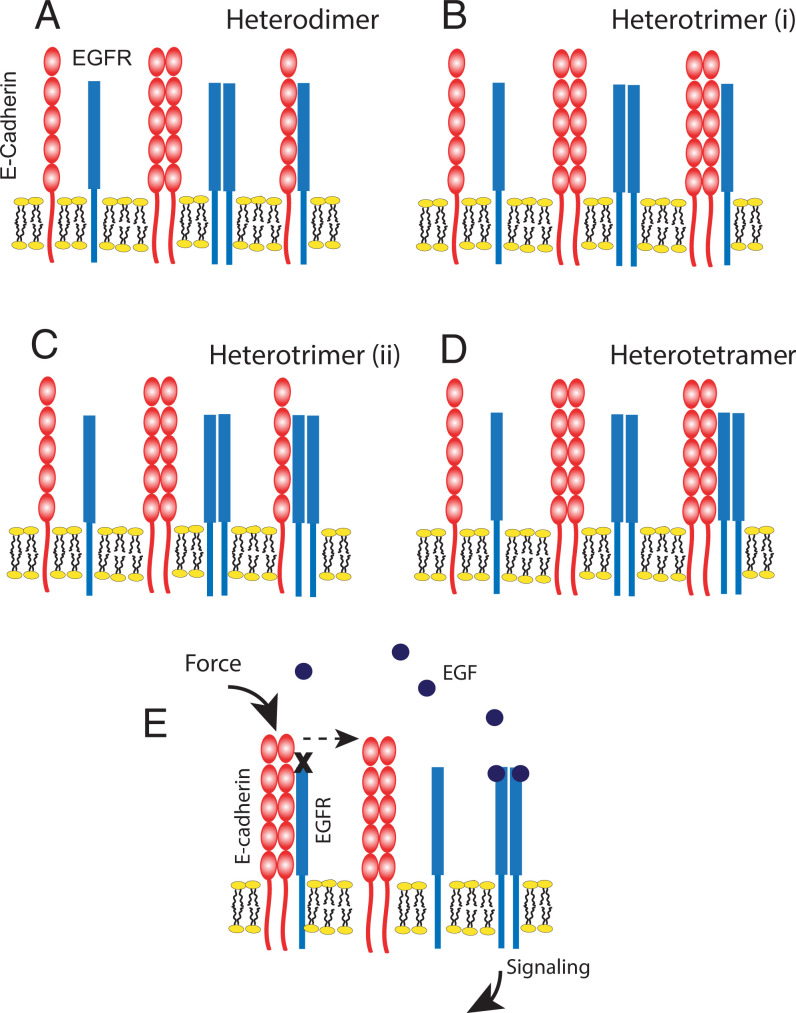
Increased intercellular tension is associated with enhanced cell proliferation and tissue growth. Here, we present evidence for a force-transduction mechanism that links mechanical perturbations of epithelial (E)-cadherin (CDH1) receptors to the force-dependent activation of epidermal growth factor receptor (EGFR, ERBB1)-a key regulator of cell proliferation. Here, coimmunoprecipitation studies first show that E-cadherin and EGFR form complexes at the plasma membrane that are disrupted by either epidermal growth factor (EGF) or increased tension on homophilic E-cadherin bonds. Although force on E-cadherin bonds disrupts the complex in the absence of EGF, soluble EGF is required to mechanically activate EGFR at cadherin adhesions. Fully quantified spectral imaging fluorescence resonance energy transfer further revealed that E-cadherin and EGFR directly associate to form a heterotrimeric complex of two cadherins and one EGFR protein. Together, these results support a model in which the tugging forces on homophilic E-cadherin bonds trigger force-activated signaling by releasing EGFR monomers to dimerize, bind EGF ligand, and signal. These findings reveal the initial steps in E-cadherin-mediated force transduction that directly link intercellular force fluctuations to the activation of growth regulatory signaling cascades.
Keywords: FRET; MAPK; cadherin; epidermal growth factor receptor; mechanotransduction.
Copyright © 2022 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Marsden M., DeSimone D. W., Integrin-ECM interactions regulate cadherin-dependent cell adhesion and are required for convergent extension in Xenopus. Curr. Biol. 13, 1182–1191 (2003). - PubMed
-
- Hayashi T., Carthew R. W., Surface mechanics mediate pattern formation in the developing retina. Nature 431, 647–652 (2004). - PubMed
-
- Tzima E., et al. , A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
