

A simple guide to de novo transcriptome assembly and annotation
- PMID: 35076693
- PMCID: PMC8921630
- DOI: 10.1093/bib/bbab563
A simple guide to de novo transcriptome assembly and annotation
Abstract
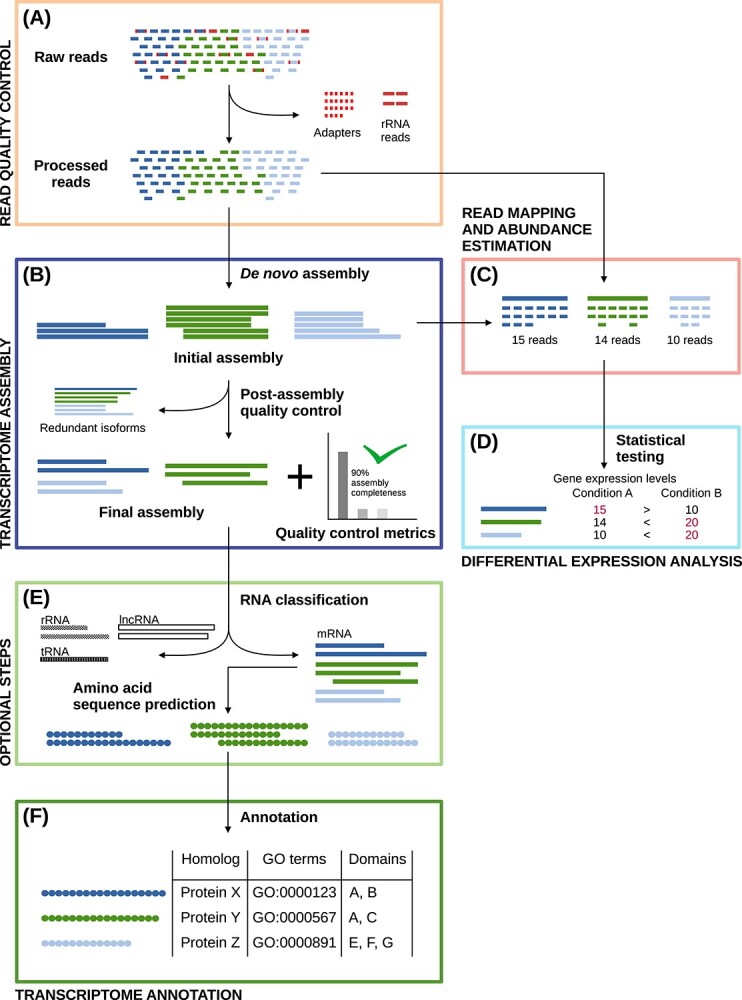
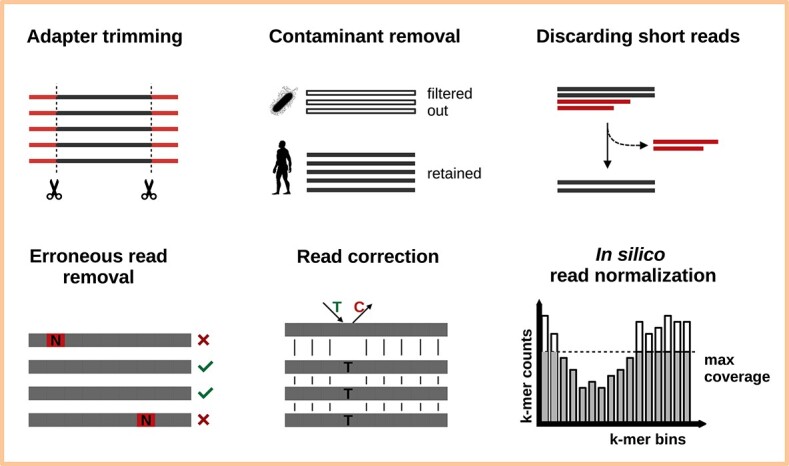
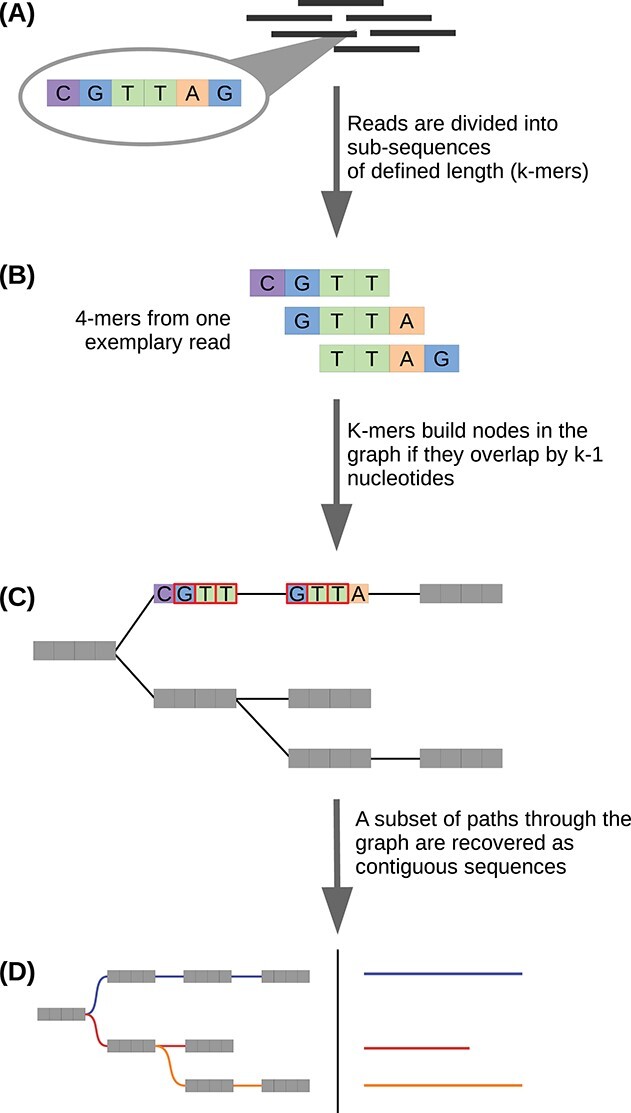
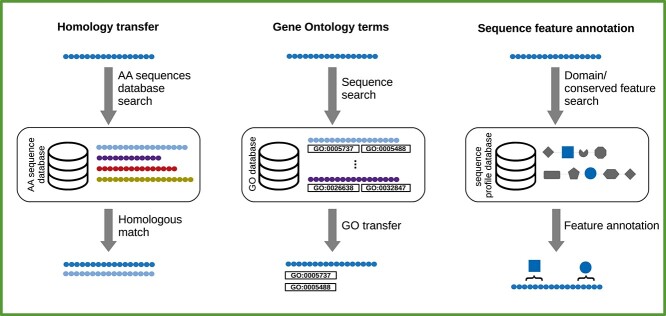
A transcriptome constructed from short-read RNA sequencing (RNA-seq) is an easily attainable proxy catalog of protein-coding genes when genome assembly is unnecessary, expensive or difficult. In the absence of a sequenced genome to guide the reconstruction process, the transcriptome must be assembled de novo using only the information available in the RNA-seq reads. Subsequently, the sequences must be annotated in order to identify sequence-intrinsic and evolutionary features in them (for example, protein-coding regions). Although straightforward at first glance, de novo transcriptome assembly and annotation can quickly prove to be challenging undertakings. In addition to familiarizing themselves with the conceptual and technical intricacies of the tasks at hand and the numerous pre- and post-processing steps involved, those interested must also grapple with an overwhelmingly large choice of tools. The lack of standardized workflows, fast pace of development of new tools and techniques and paucity of authoritative literature have served to exacerbate the difficulty of the task even further. Here, we present a comprehensive overview of de novo transcriptome assembly and annotation. We discuss the procedures involved, including pre- and post-processing steps, and present a compendium of corresponding tools.
Keywords: RNA-seq; annotation; assembly; de novo; tools; transcriptome.
© The Author(s) 2022. Published by Oxford University Press.
Figures
References
-
- Buccitelli C, Selbach M. mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet October 2020;21(10):630–44. - PubMed
-
- Schimmel P. The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nat Rev Mol Cell Biol January 2018;19(1):45–58. - PubMed
