

Efficacy and Safety of Vamorolone in Duchenne Muscular Dystrophy: A 30-Month Nonrandomized Controlled Open-Label Extension Trial
- PMID: 35076703
- PMCID: PMC8790668
- DOI: 10.1001/jamanetworkopen.2021.44178
Efficacy and Safety of Vamorolone in Duchenne Muscular Dystrophy: A 30-Month Nonrandomized Controlled Open-Label Extension Trial
Abstract
Importance: Vamorolone is a synthetic steroidal drug with potent anti-inflammatory properties. Initial open-label, multiple ascending dose-finding studies of vamorolone among boys with Duchenne muscular dystrophy (DMD) found significant motor function improvement after 6 months treatment in higher-dose (ie, ≥2.0 mg/kg/d) groups.
Objective: To investigate outcomes after 30 months of open-label vamorolone treatment.
Design, setting, and participants: This nonrandomized controlled trial was conducted by the Cooperative International Neuromuscular Research Group at 11 US and non-US study sites. Participants were 46 boys ages 4.5 to 7.5 years with DMD who completed the 6-month dose-finding study. Data were analyzed from July 2020 through November 2021.
Interventions: Participants were enrolled in a 24-month, long-term extension (LTE) study with vamorolone dose escalated to 2.0 or 6.0 mg/kg/d.
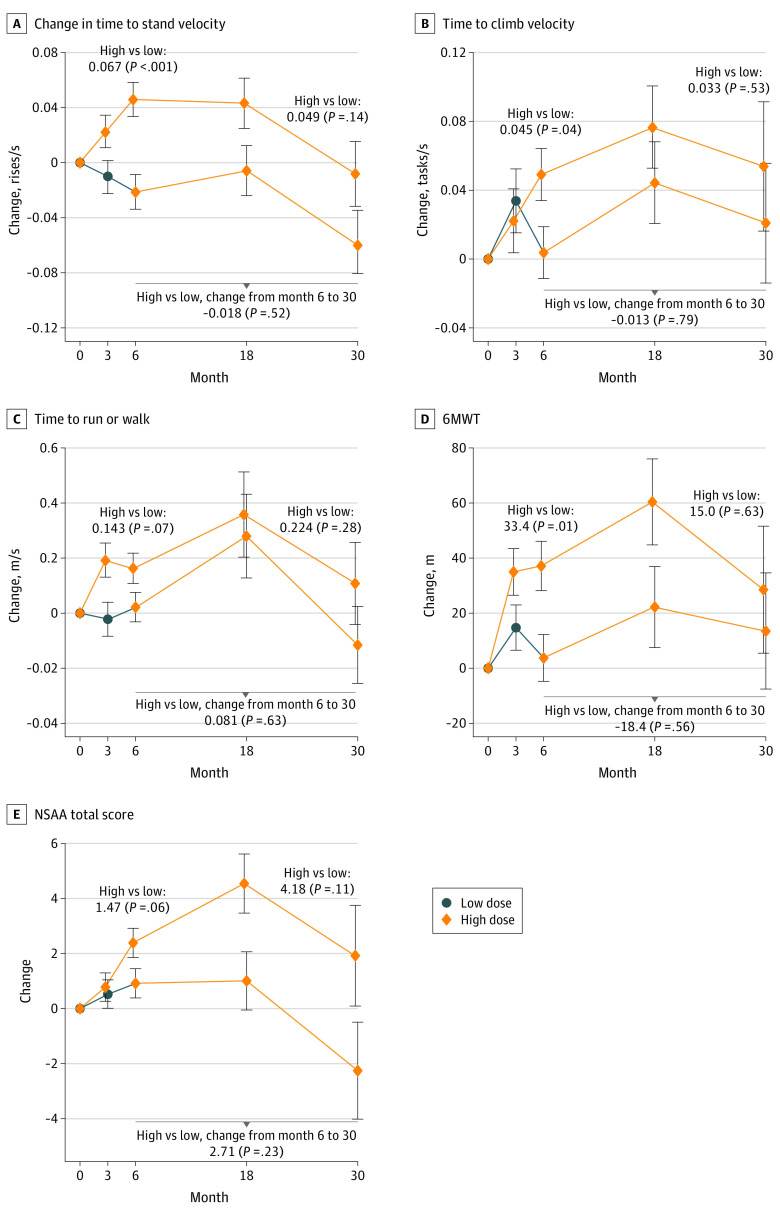
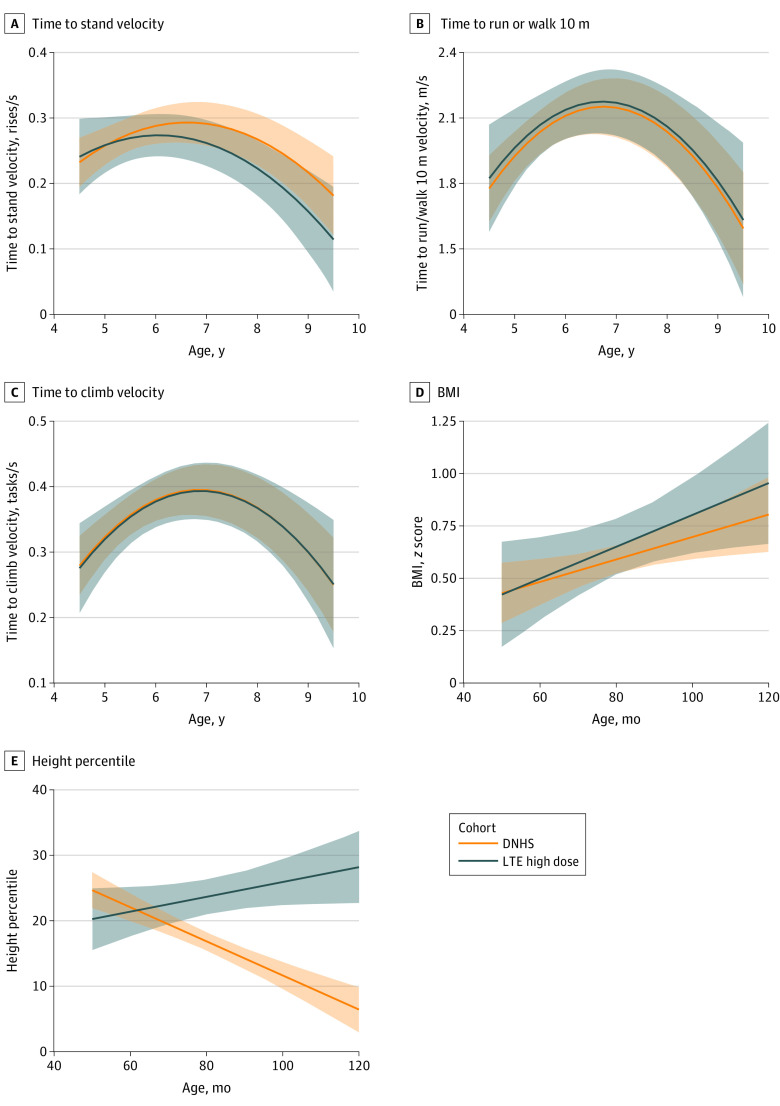
Main outcomes and measures: Change in time-to-stand (TTSTAND) velocity from dose-finding baseline to end of LTE study was the primary outcome. Efficacy assessments included timed function tests, 6-minute walk test, and NorthStar Ambulatory Assessment (NSAA). Participants with DMD treated with glucocorticoids from the Duchenne Natural History Study (DNHS) and NorthStar United Kingdom (NSUK) Network were matched and compared with participants in the LTE study receiving higher doses of vamorolone.
Results: Among 46 boys with DMD who completed the dose-finding study, 41 boys (mean [SD] age, 5.33 [0.96] years) completed the LTE study. Among 21 participants treated with higher-dose (ie, ≥2.0 mg/kg/d) vamorolone consistently throughout the 6-month dose-finding and 24-month LTE studies with data available at 30 months, there was a decrease in mean (SD) TTSTAND velocity from baseline to 30 months (0.206 [0.070] rises/s vs 0.189 (0.124) rises/s), which was not a statistically significant change (-0.011 rises/s; CI, -0.068 to 0.046 rises/s). There were no statistically significant differences between participants receiving higher-dose vamorolone and matched participants in the historical control groups receiving glucocorticoid treatment (75 patients in DNHS and 110 patients in NSUK) over a 2-year period in NSAA total score change (0.22 units vs NSUK; 95% CI, -4.48 to 4.04]; P = .92), body mass index z score change (0.002 vs DNHS SD/mo; 95% CI, -0.006 to 0.010; P = .58), or timed function test change. Vamorolone at doses up to 6.0 mg/kg/d was well tolerated, with 5 of 46 participants discontinuing prematurely and for reasons not associated with study drug. Participants in the DNHS treated with glucocorticoids had significant growth delay in comparison with participants treated with vamorolone who had stable height percentiles (0.37 percentile/mo; 95% CI, 0.23 to 0.52 percentile/mo) over time.
Conclusions and relevance: This study found that vamorolone treatment was not associated with a change in TTSTAND velocity from baseline to 30 months among boys with DMD aged 4 to 7 years at enrollment. Vamorolone was associated with maintenance of muscle strength and function up to 30 months, similar to standard of care glucocorticoid therapy, and improved height velocity compared with growth deceleration associated with glucocorticoid treatment, suggesting that vamorolone may be an attractive candidate for treatment of DMD.
Trial registration: ClinicalTrials.gov Identifier: NCT03038399.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
