

Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration
- PMID: 35078537
- PMCID: PMC8788129
- DOI: 10.1186/s40035-021-00278-7
Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration
Abstract
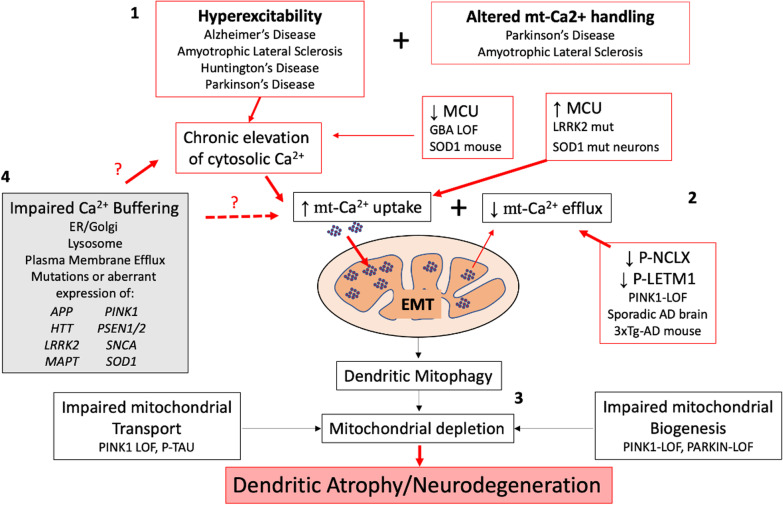
Glutamate is the most commonly engaged neurotransmitter in the mammalian central nervous system, acting to mediate excitatory neurotransmission. However, high levels of glutamatergic input elicit excitotoxicity, contributing to neuronal cell death following acute brain injuries such as stroke and trauma. While excitotoxic cell death has also been implicated in some neurodegenerative disease models, the role of acute apoptotic cell death remains controversial in the setting of chronic neurodegeneration. Nevertheless, it is clear that excitatory synaptic dysregulation contributes to neurodegeneration, as evidenced by protective effects of partial N-methyl-D-aspartate receptor antagonists. Here, we review evidence for sublethal excitatory injuries in relation to neurodegeneration associated with Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis and Huntington's disease. In contrast to classic excitotoxicity, emerging evidence implicates dysregulation of mitochondrial calcium handling in excitatory post-synaptic neurodegeneration. We discuss mechanisms that regulate mitochondrial calcium uptake and release, the impact of LRRK2, PINK1, Parkin, beta-amyloid and glucocerebrosidase on mitochondrial calcium transporters, and the role of autophagic mitochondrial loss in axodendritic shrinkage. Finally, we discuss strategies for normalizing the flux of calcium into and out of the mitochondrial matrix, thereby preventing mitochondrial calcium toxicity and excitotoxic dendritic loss. While the mechanisms that underlie increased uptake or decreased release of mitochondrial calcium vary in different model systems, a common set of strategies to normalize mitochondrial calcium flux can prevent excitatory mitochondrial toxicity and may be neuroprotective in multiple disease contexts.
Keywords: Alzheimer’s disease; Amyotrophic lateral sclerosis; Beta-amyloid; Excitotoxicity; Glucocerebrosidase; Huntington’s disease; LRRK2; Mitochondrial calcium; Mitochondrial calcium uniporter; Mitophagy; NCLX antiporter; PINK1; Parkinson’s disease.
© 2022. The Author(s).
Conflict of interest statement
CTC is named on a patent application targeting FBX07 (PCT/US2018/039327) submitted jointly by the University of Pittsburgh and the US Department of Veterans Affairs. The other authors declare that they have no competing interests.
Figures
References
-
- Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience. 1995;68(3):615–619. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
