

A genome-wide association study of serum proteins reveals shared loci with common diseases
- PMID: 35078996
- PMCID: PMC8789779
- DOI: 10.1038/s41467-021-27850-z
A genome-wide association study of serum proteins reveals shared loci with common diseases
Abstract
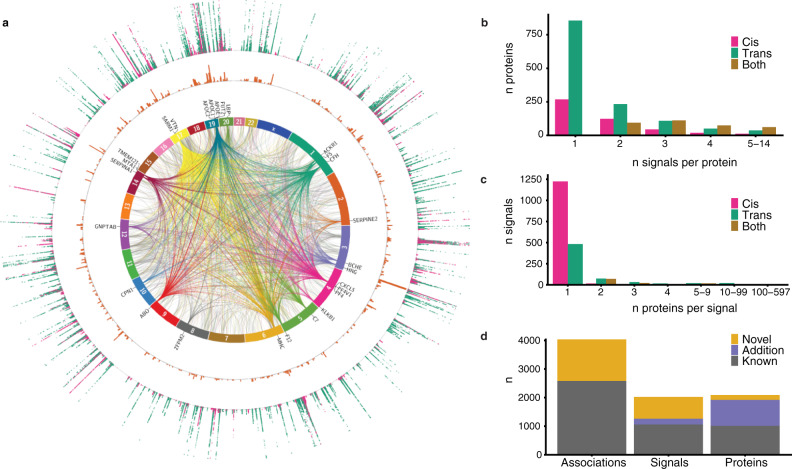
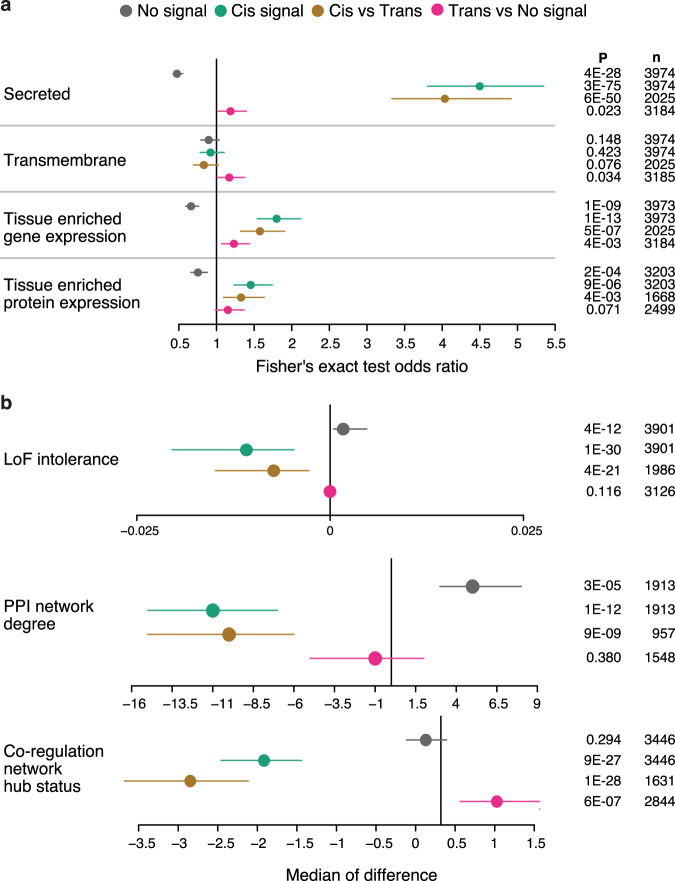
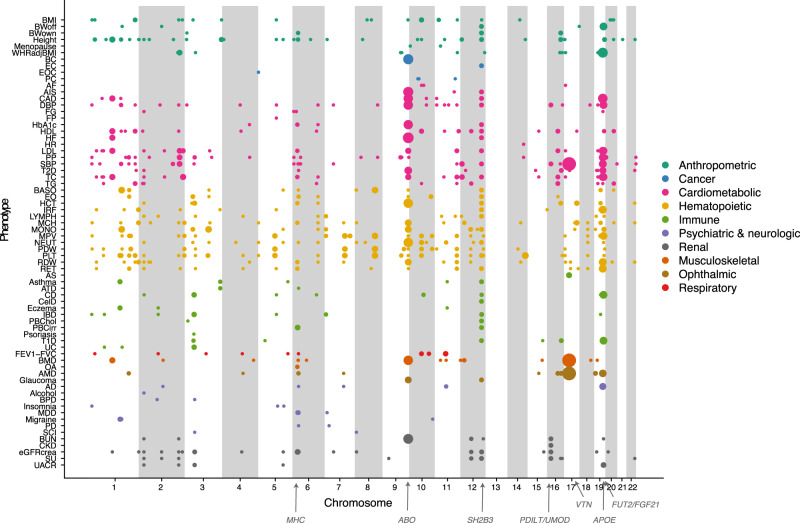
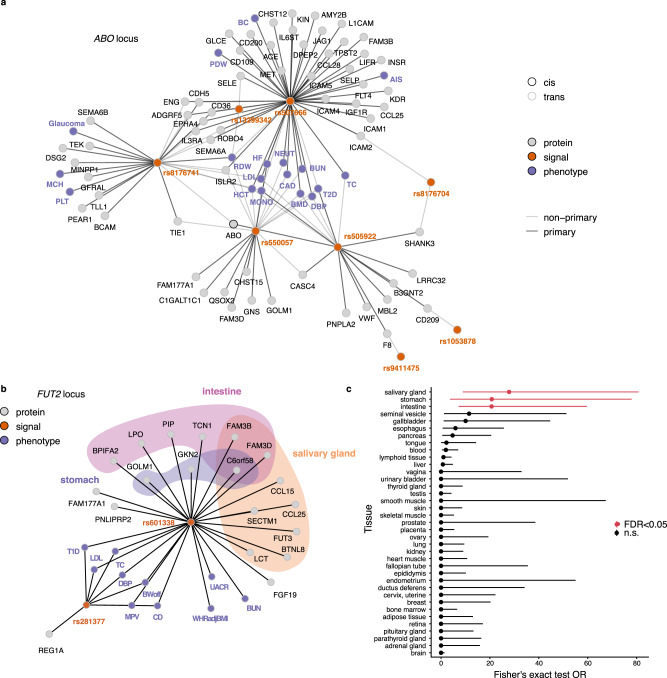
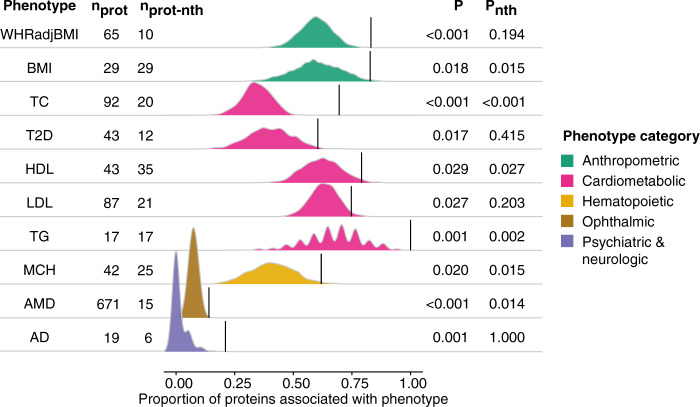
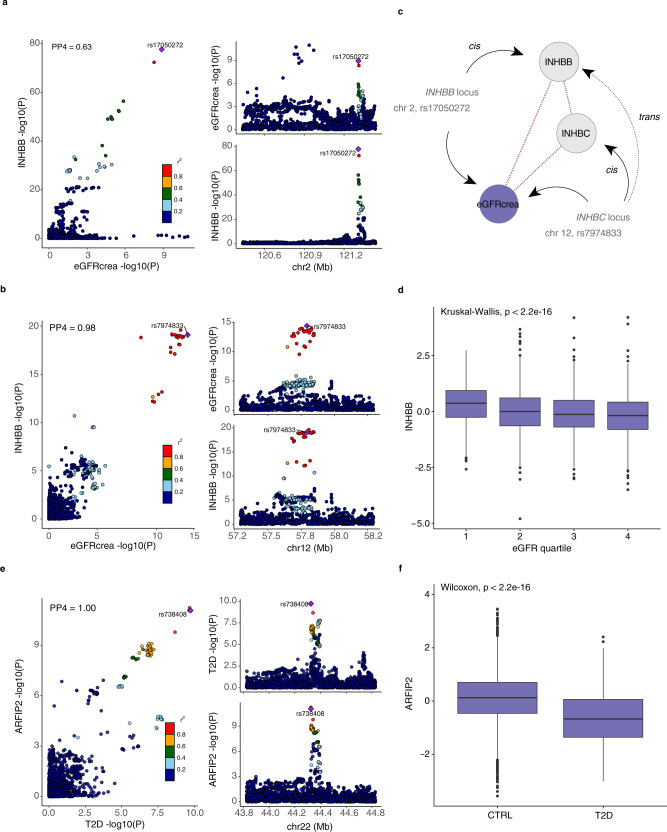
With the growing number of genetic association studies, the genotype-phenotype atlas has become increasingly more complex, yet the functional consequences of most disease associated alleles is not understood. The measurement of protein level variation in solid tissues and biofluids integrated with genetic variants offers a path to deeper functional insights. Here we present a large-scale proteogenomic study in 5,368 individuals, revealing 4,035 independent associations between genetic variants and 2,091 serum proteins, of which 36% are previously unreported. The majority of both cis- and trans-acting genetic signals are unique for a single protein, although our results also highlight numerous highly pleiotropic genetic effects on protein levels and demonstrate that a protein's genetic association profile reflects certain characteristics of the protein, including its location in protein networks, tissue specificity and intolerance to loss of function mutations. Integrating protein measurements with deep phenotyping of the cohort, we observe substantial enrichment of phenotype associations for serum proteins regulated by established GWAS loci, and offer new insights into the interplay between genetics, serum protein levels and complex disease.
© 2022. The Author(s).
Conflict of interest statement
The study was supported by the Novartis Institute for Biomedical Research, and protein measurements for the AGES-Reykjavik cohort were performed at SomaLogic. J.R.L. and L.L.J. are employees and stockholders of Novartis. All other authors have no conflict of interests to declare.
Figures
References
-
- Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
