

Mitochondrial-nuclear epistasis underlying phenotypic variation in breast cancer pathology
- PMID: 35082309
- PMCID: PMC8791930
- DOI: 10.1038/s41598-022-05148-4
Mitochondrial-nuclear epistasis underlying phenotypic variation in breast cancer pathology
Abstract
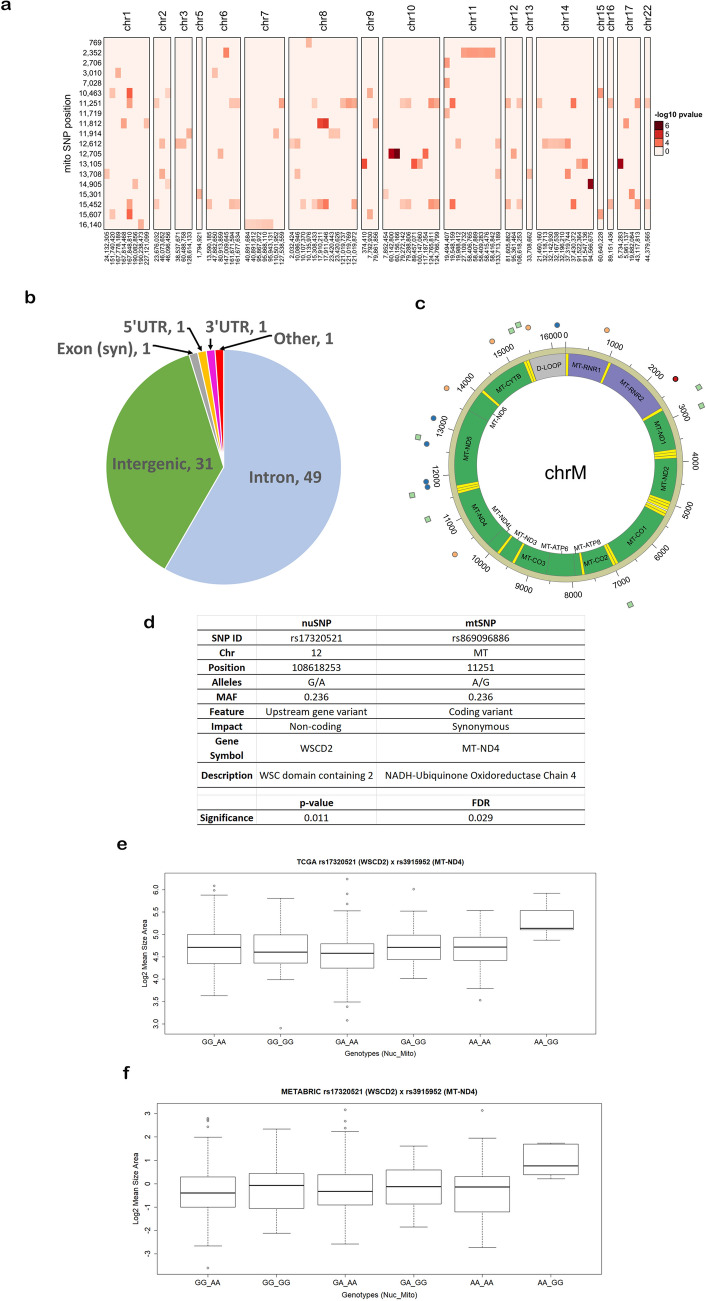
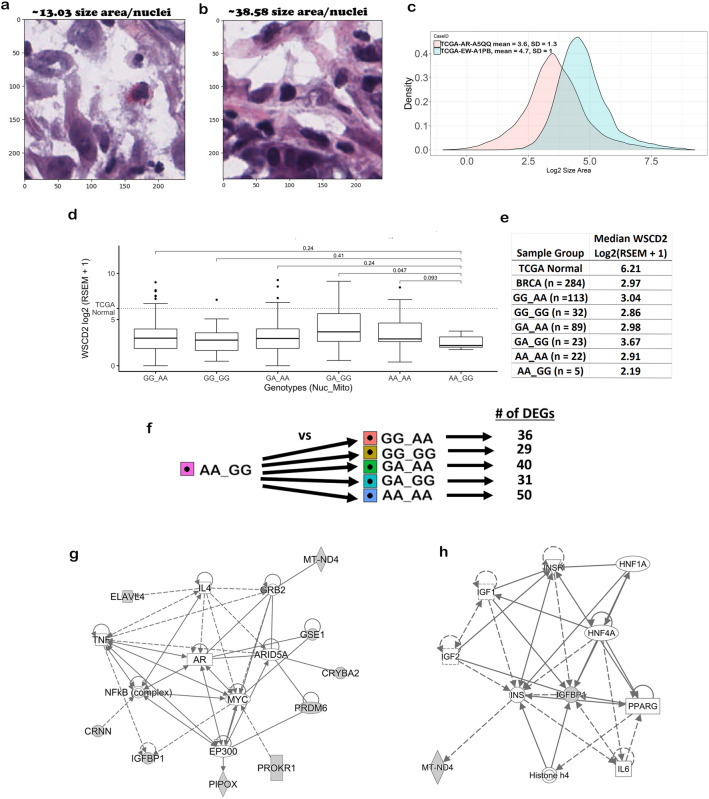
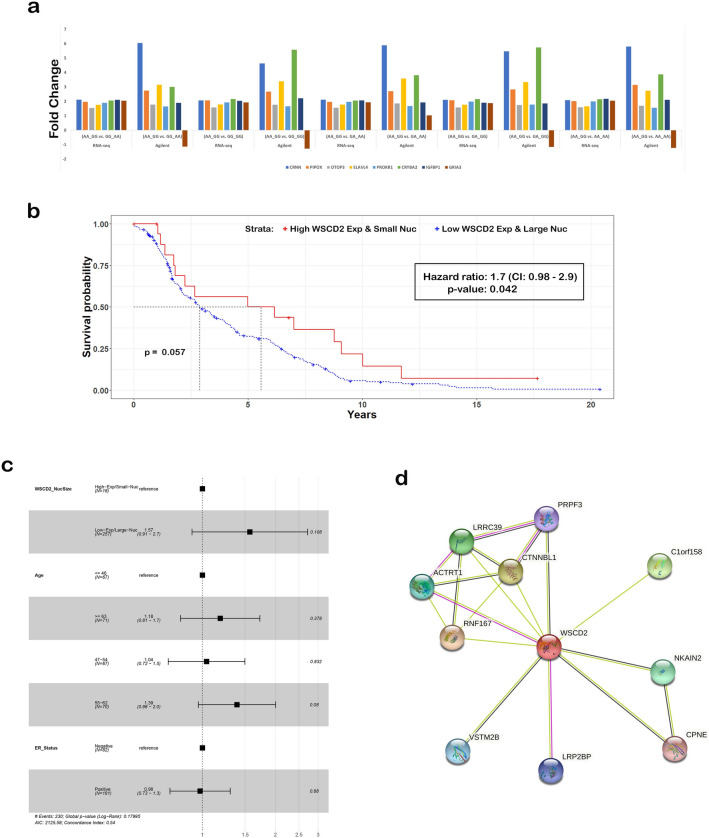
The interplay between genes harboring single nucleotide polymorphisms (SNPs) is vital to better understand underlying contributions to the etiology of breast cancer. Much attention has been paid to epistasis between nuclear genes or mutations in the mitochondrial genome. However, there is limited understanding about the epistatic effects of genetic variants in the nuclear and mitochondrial genomes jointly on breast cancer. We tested the interaction of germline SNPs in the mitochondrial (mtSNPs) and nuclear (nuSNPs) genomes of female breast cancer patients in The Cancer Genome Atlas (TCGA) for association with morphological features extracted from hematoxylin and eosin (H&E)-stained pathology images. We identified 115 significant (q-value < 0.05) mito-nuclear interactions that increased nuclei size by as much as 12%. One interaction between nuSNP rs17320521 in an intron of the WSC Domain Containing 2 (WSCD2) gene and mtSNP rs869096886, a synonymous variant mapped to the mitochondrially-encoded NADH dehydrogenase 4 (MT-ND4) gene, was confirmed in an independent breast cancer data set from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC). None of the 10 mito-nuclear interactions identified from non-diseased female breast tissues from the Genotype-Expression (GTEx) project resulted in an increase in nuclei size. Comparisons of gene expression data from the TCGA breast cancer patients with the genotype homozygous for the minor alleles of the SNPs in WSCD2 and MT-ND4 versus the other genotypes revealed core transcriptional regulator interactions and an association with insulin. Finally, a Cox proportional hazards ratio = 1.7 (C.I. 0.98-2.9, p-value = 0.042) and Kaplan-Meier plot suggest that the TCGA female breast cancer patients with low gene expression of WSCD2 coupled with large nuclei have an increased risk of mortality. The intergenomic dependency between the two variants may constitute an inherent susceptibility of a more severe form of breast cancer and points to genetic targets for further investigation of additional determinants of the disease.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
- R01 DA006227/DA/NIDA NIH HHS/United States
- R01 MH101782/MH/NIMH NIH HHS/United States
- R01 MH101810/MH/NIMH NIH HHS/United States
- R01 MH101819/MH/NIMH NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- R01 MH101820/MH/NIMH NIH HHS/United States
- R01 MH101822/MH/NIMH NIH HHS/United States
- R01 MH101825/MH/NIMH NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
