

Brahma safeguards canalization of cardiac mesoderm differentiation
- PMID: 35082446
- PMCID: PMC9196993
- DOI: 10.1038/s41586-021-04336-y
Brahma safeguards canalization of cardiac mesoderm differentiation
Erratum in
-
Author Correction: Brahma safeguards canalization of cardiac mesoderm differentiation.Nature. 2025 Dec;648(8092):E2. doi: 10.1038/s41586-025-09863-6. Nature. 2025. PMID: 41254357 No abstract available.
Abstract
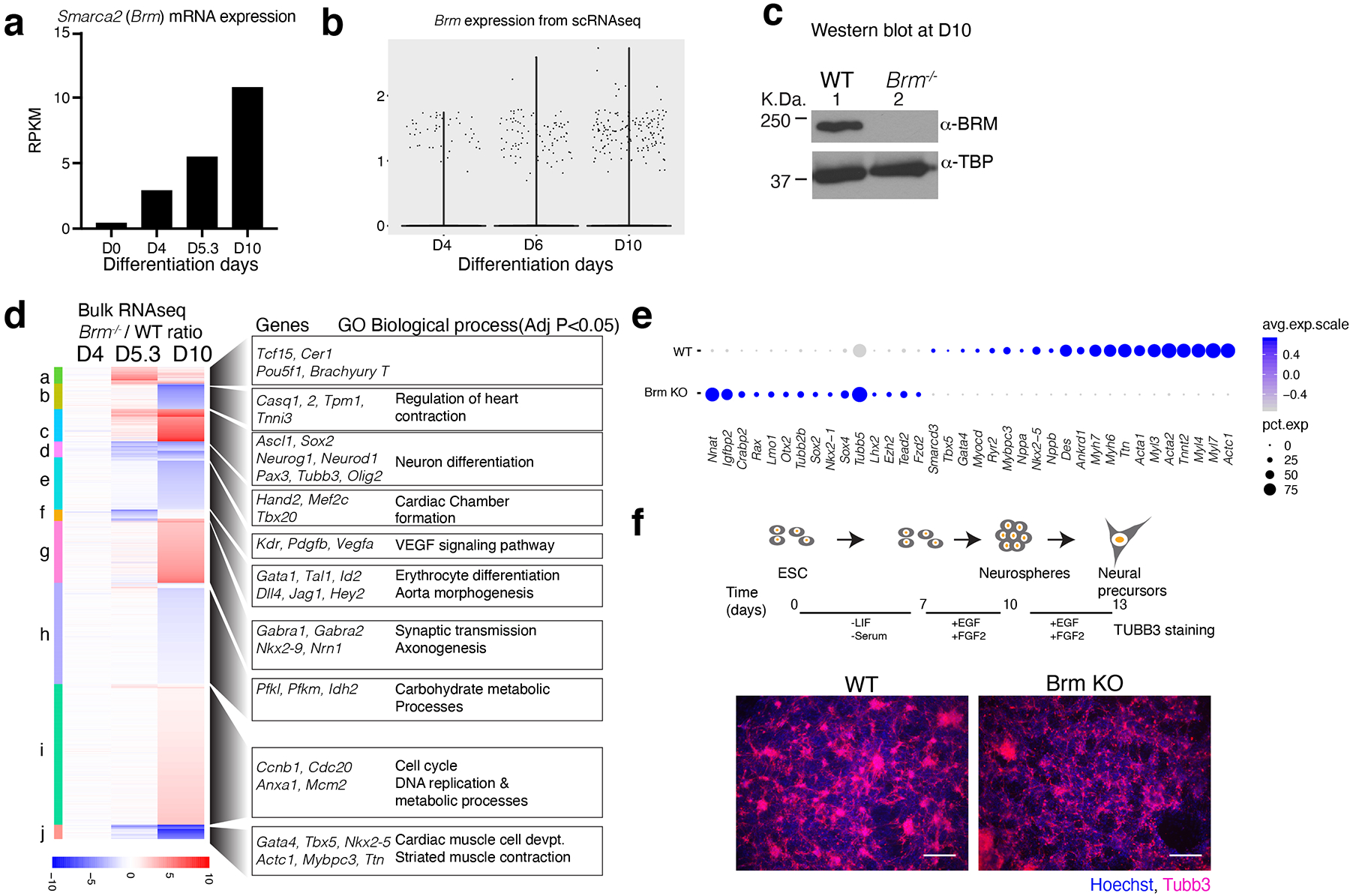
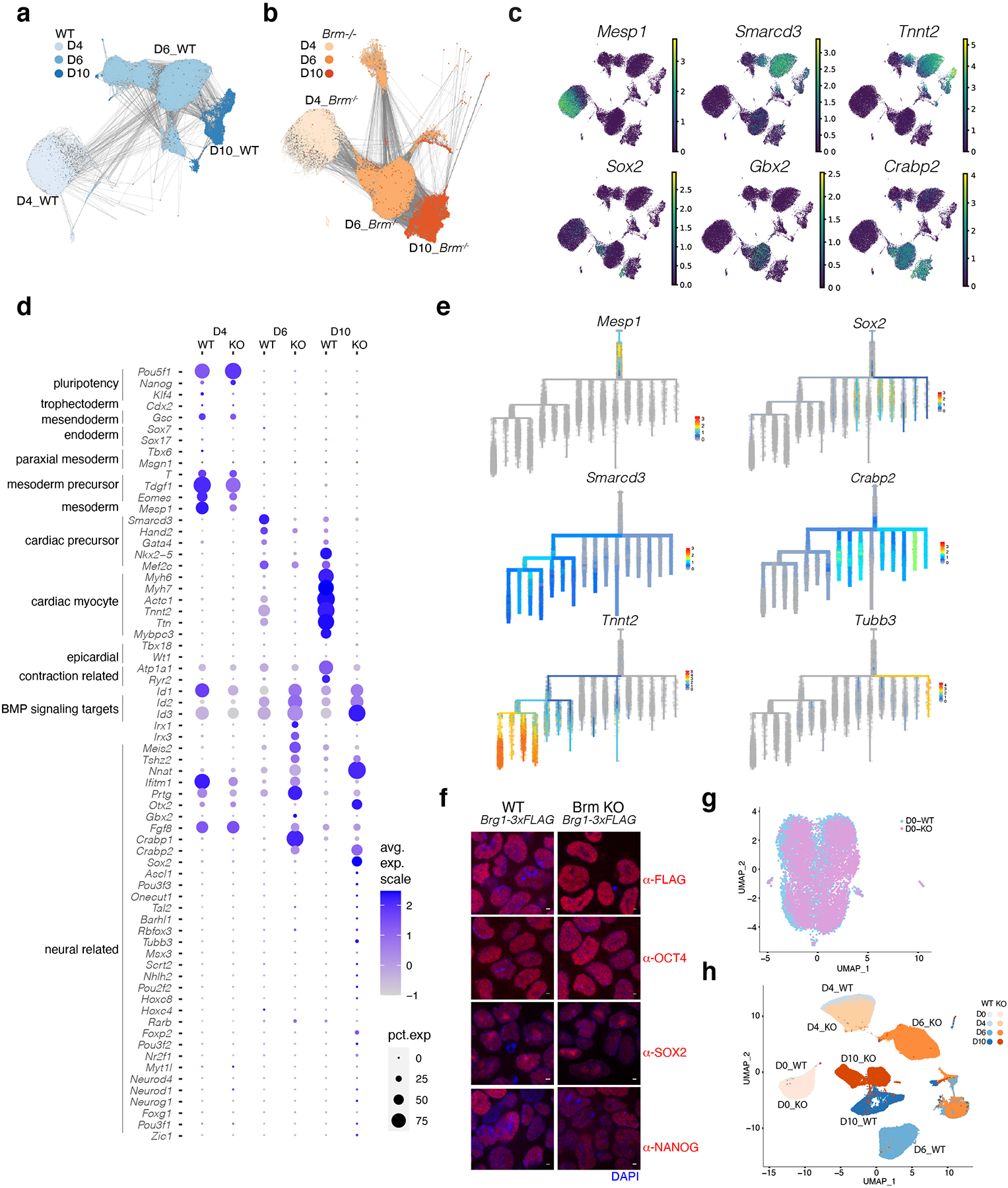
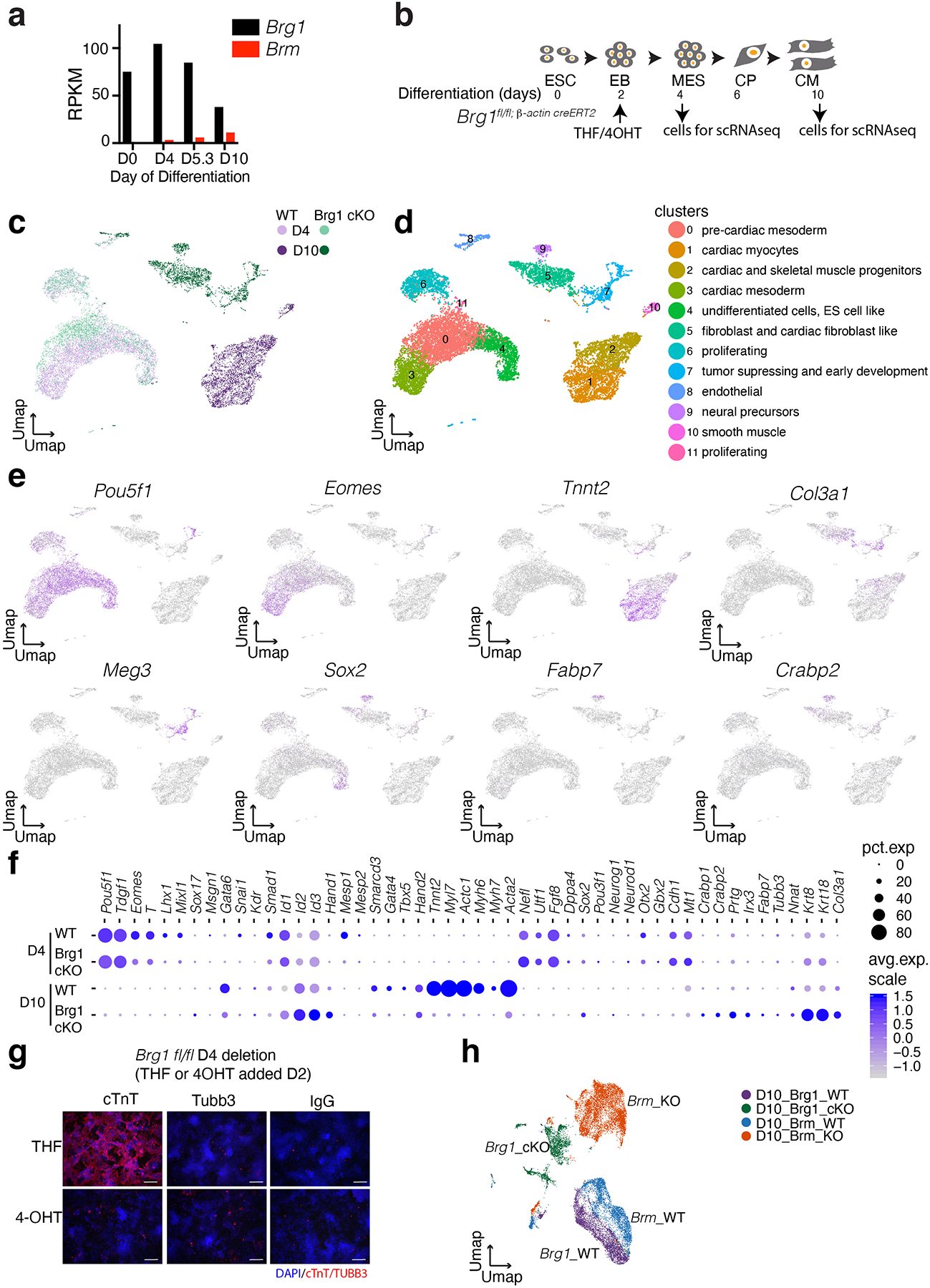
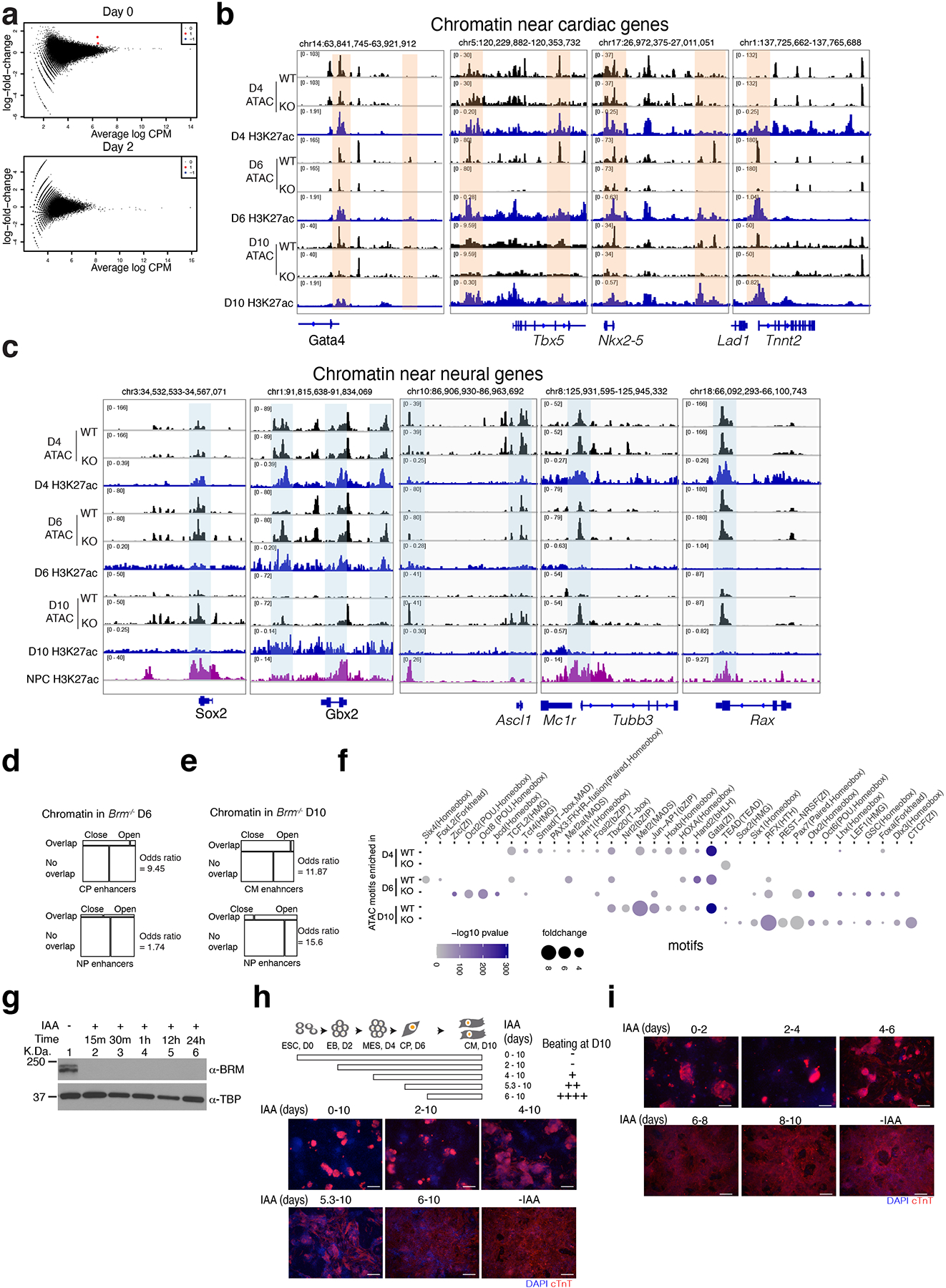
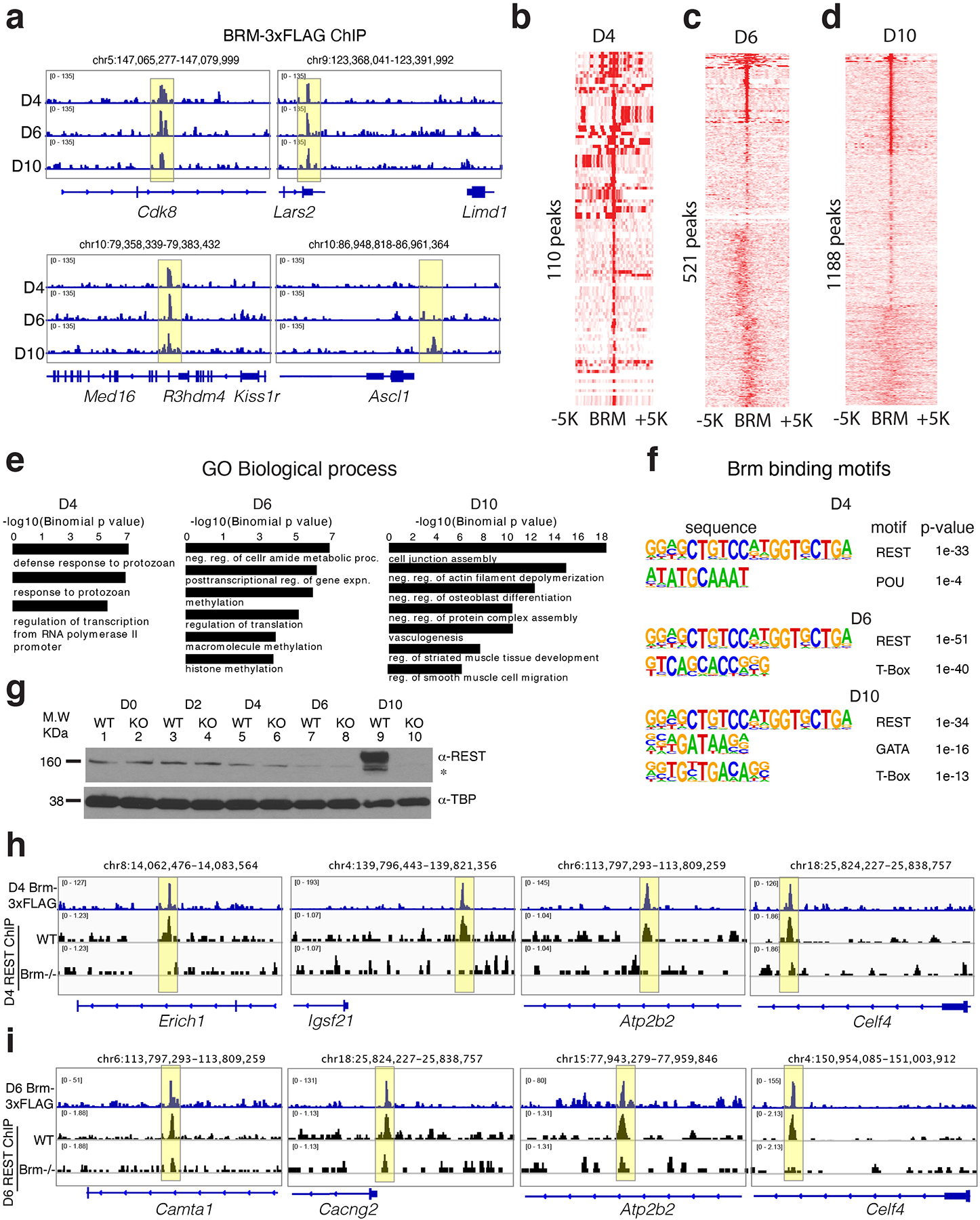
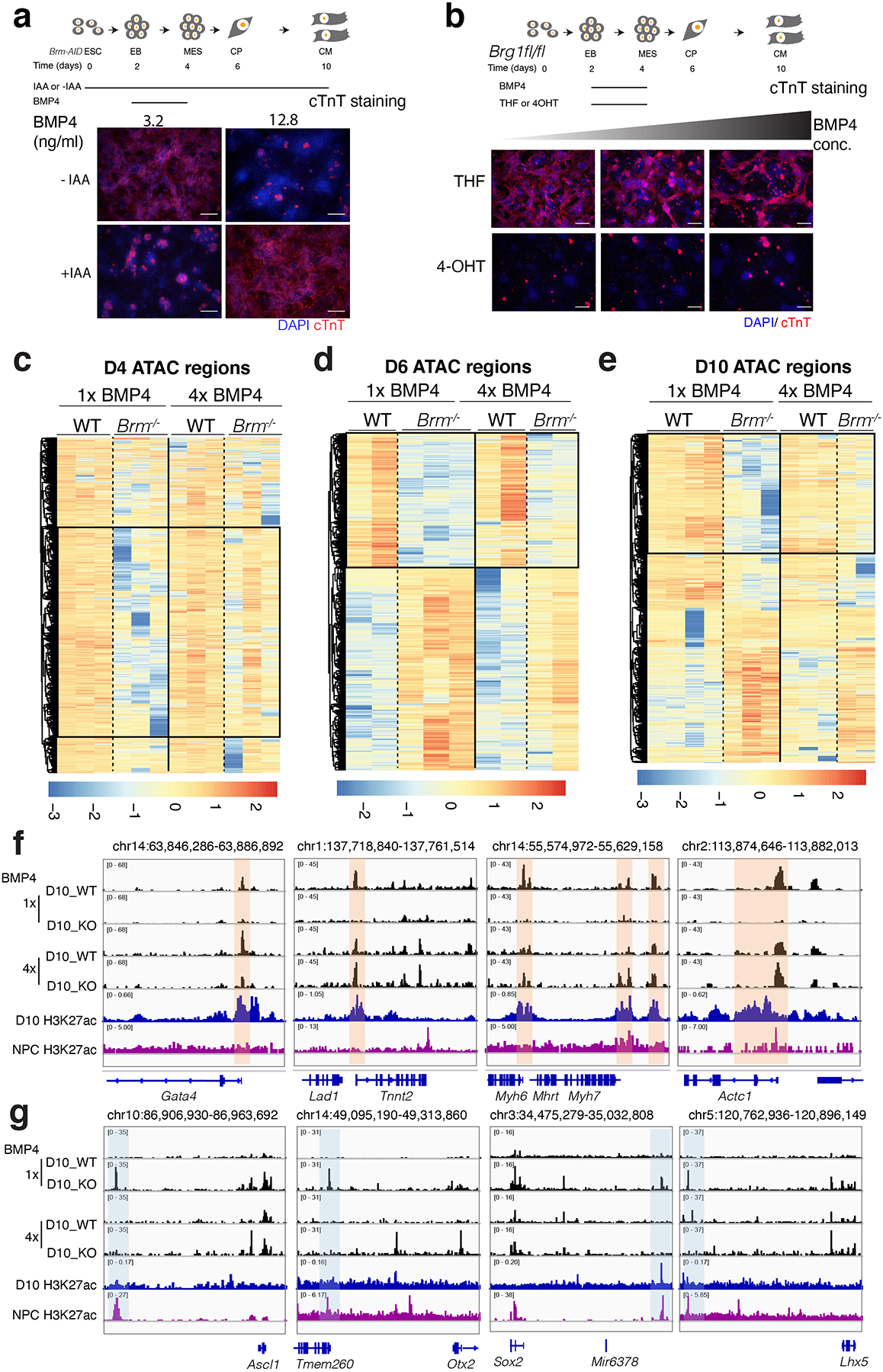
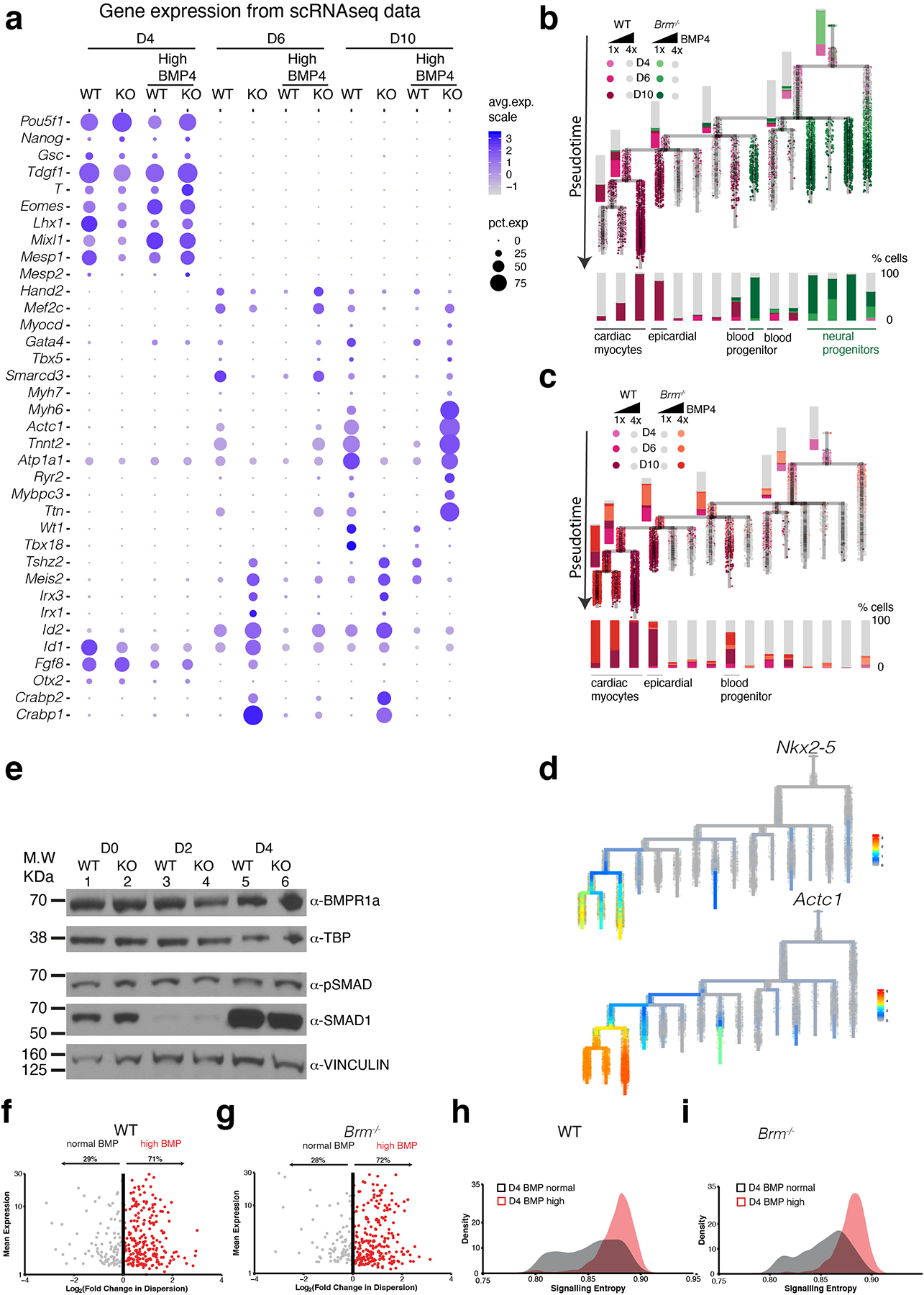
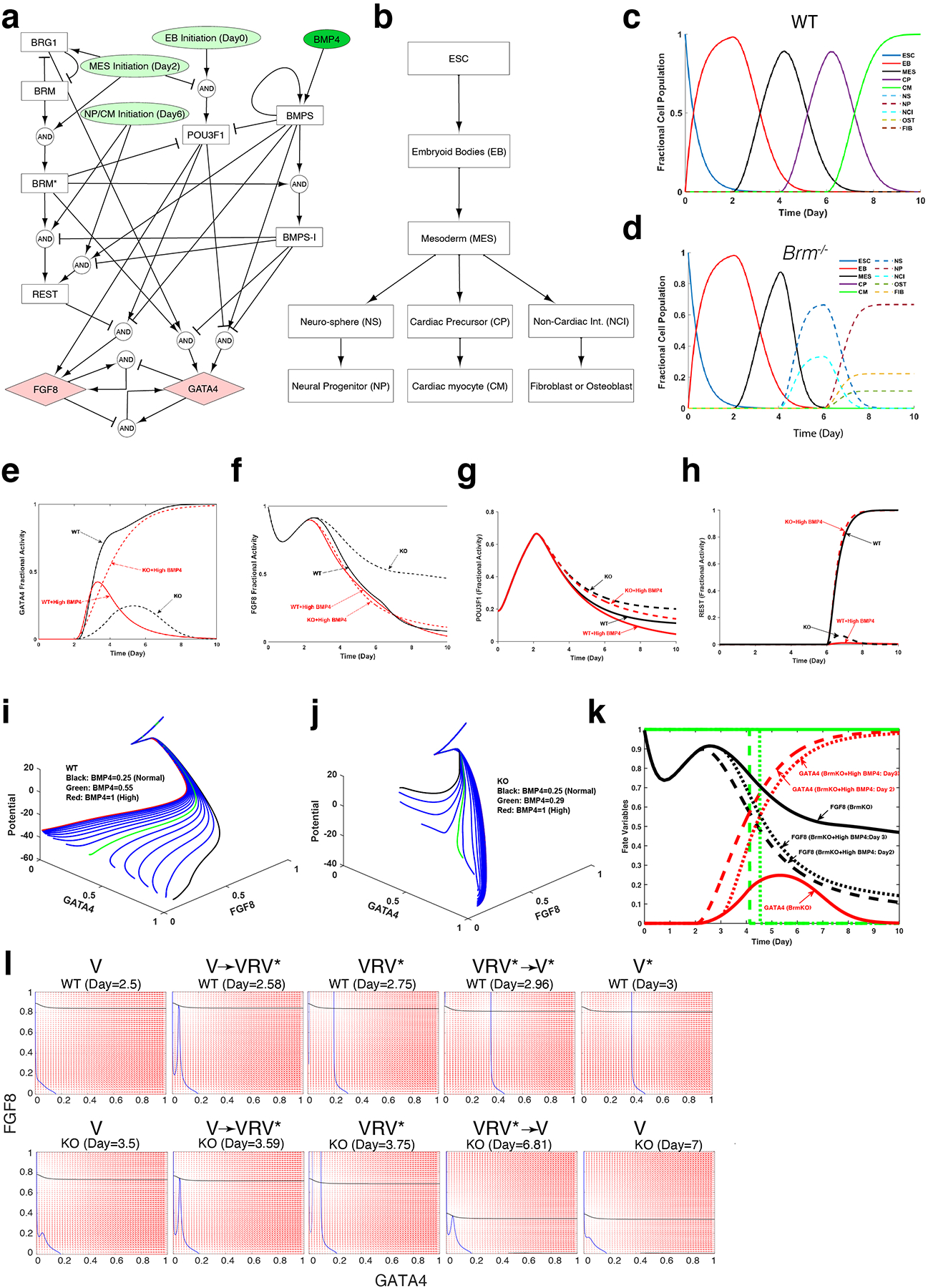
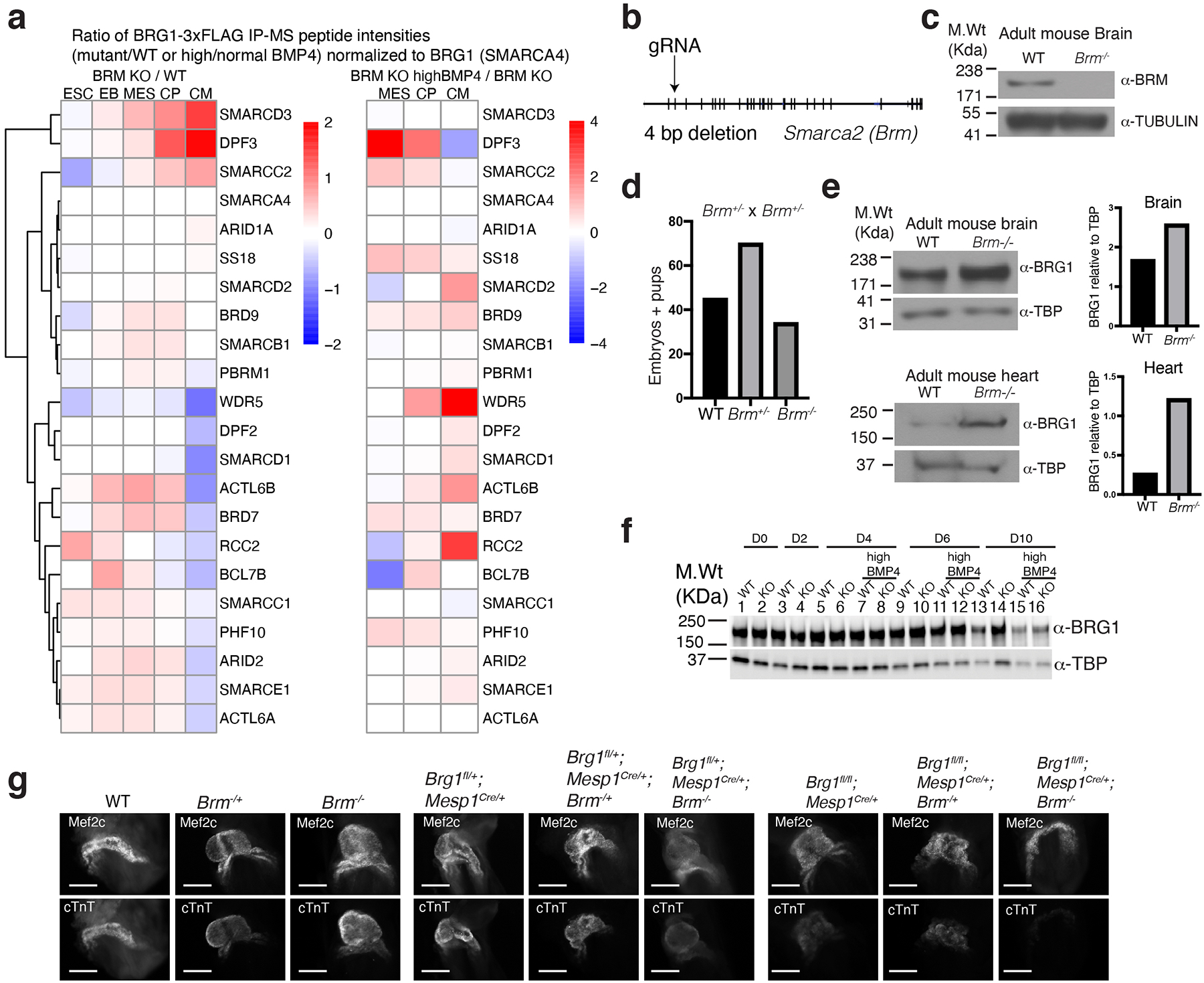
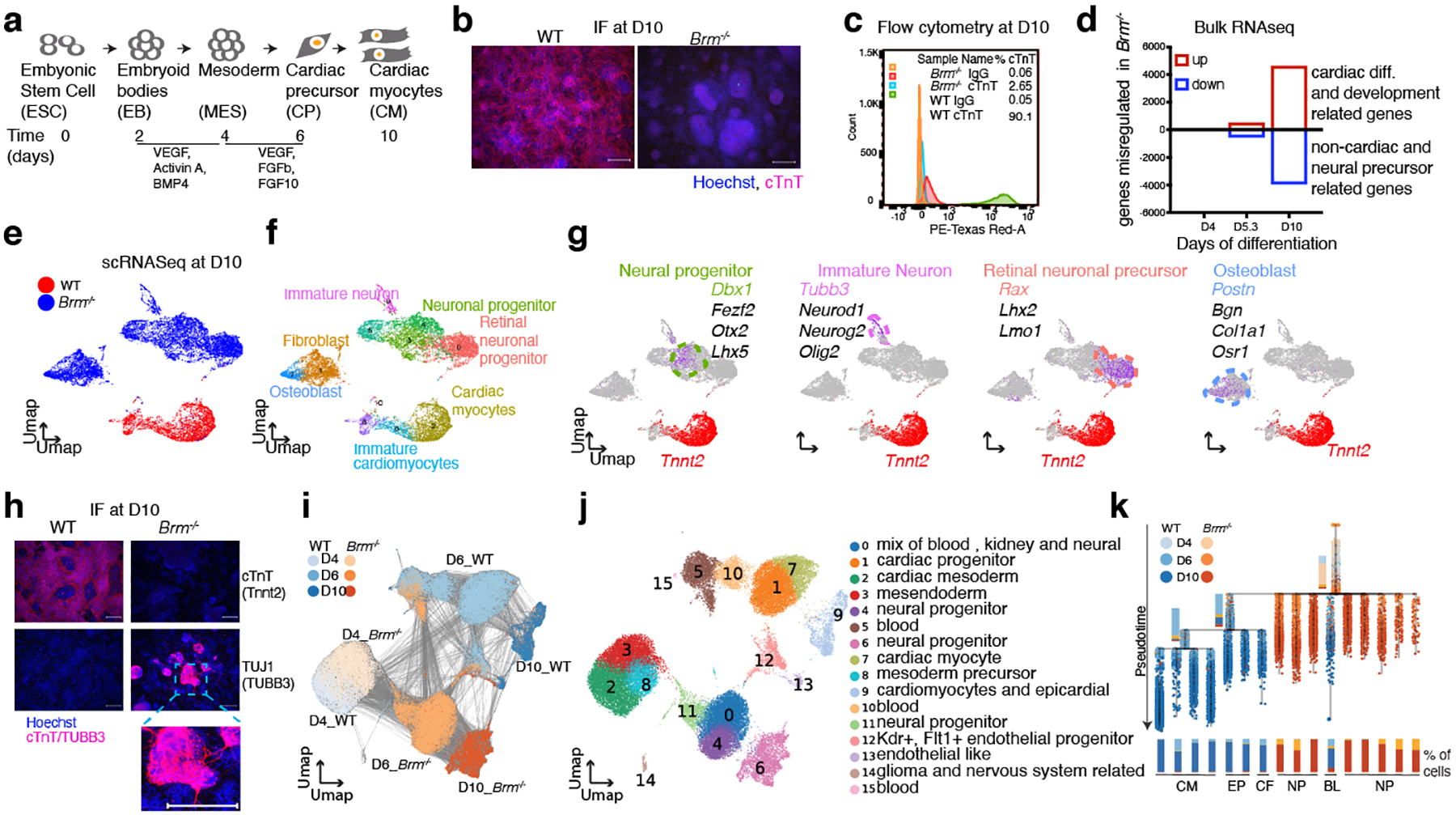
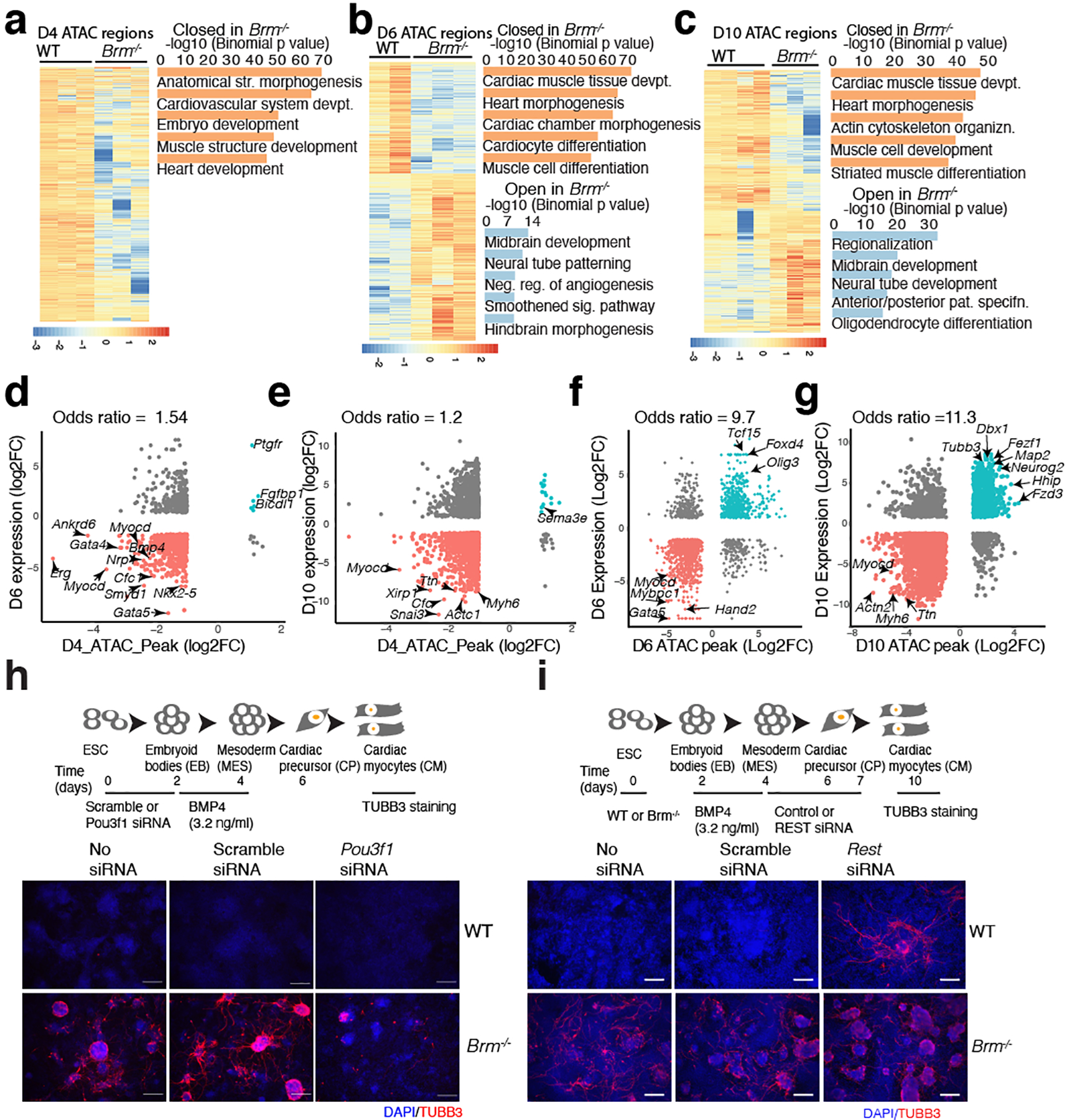
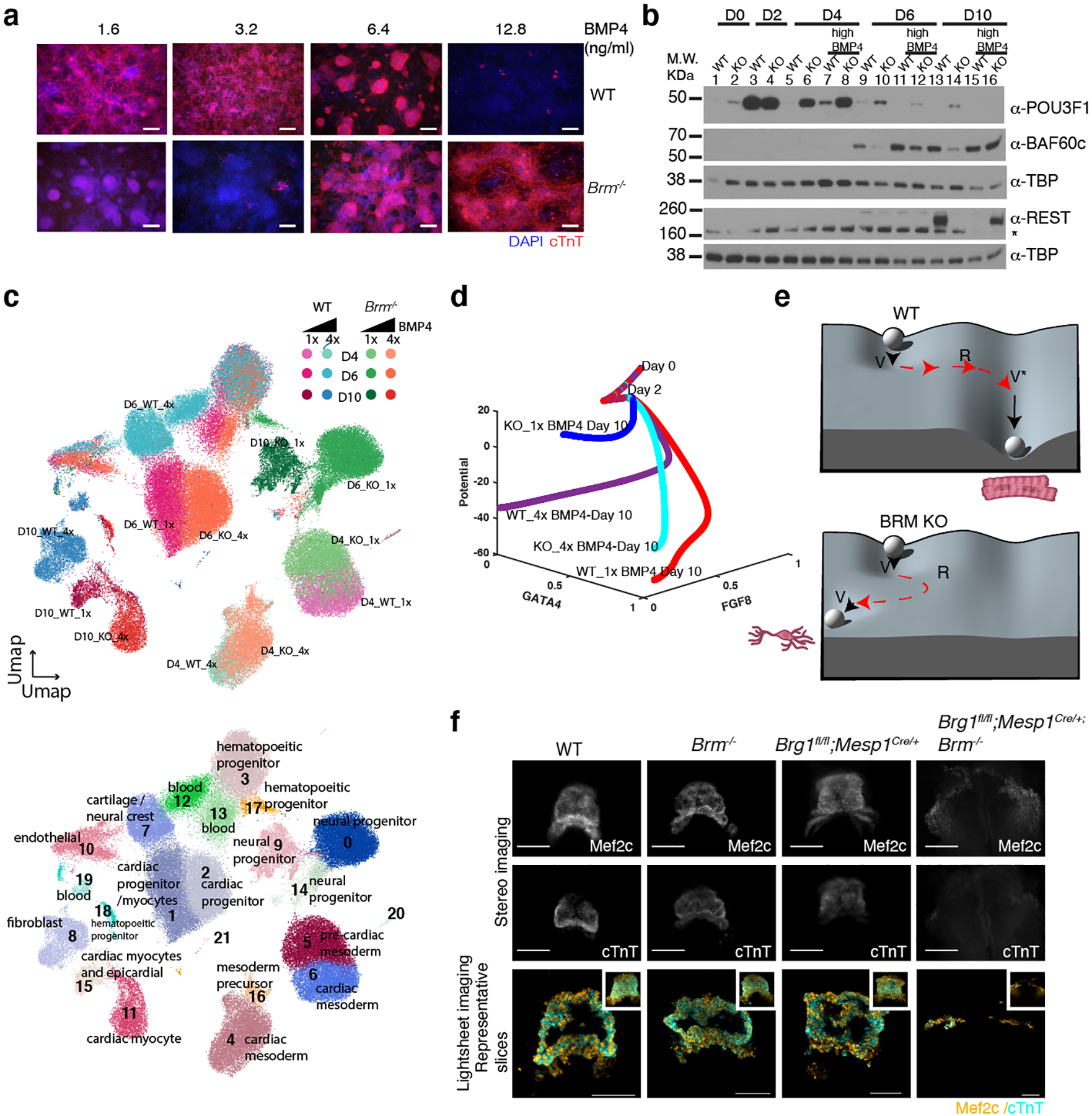
Differentiation proceeds along a continuum of increasingly fate-restricted intermediates, referred to as canalization1,2. Canalization is essential for stabilizing cell fate, but the mechanisms that underlie robust canalization are unclear. Here we show that the BRG1/BRM-associated factor (BAF) chromatin-remodelling complex ATPase gene Brm safeguards cell identity during directed cardiogenesis of mouse embryonic stem cells. Despite the establishment of a well-differentiated precardiac mesoderm, Brm-/- cells predominantly became neural precursors, violating germ layer assignment. Trajectory inference showed a sudden acquisition of a non-mesodermal identity in Brm-/- cells. Mechanistically, the loss of Brm prevented de novo accessibility of primed cardiac enhancers while increasing the expression of neurogenic factor POU3F1, preventing the binding of the neural suppressor REST and shifting the composition of BRG1 complexes. The identity switch caused by the Brm mutation was overcome by increasing BMP4 levels during mesoderm induction. Mathematical modelling supports these observations and demonstrates that Brm deletion affects cell fate trajectory by modifying saddle-node bifurcations2. In the mouse embryo, Brm deletion exacerbated mesoderm-deleted Brg1-mutant phenotypes, severely compromising cardiogenesis, and reveals an in vivo role for Brm. Our results show that Brm is a compensable safeguard of the fidelity of mesoderm chromatin states, and support a model in which developmental canalization is not a rigid irreversible path, but a highly plastic trajectory.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Figures

References
-
- Waddington CH The Strategy of the Genes, a Discussion of Some Aspects of Theoretical Biology. 20, (George Allen & Unwin Ltd, 1957).
-
- Van Houdt JKJ et al. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 44, 445–9–S1 (2012). - PubMed
-
- Tsurusaki Y et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 44, 376–378 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL114948/HL/NHLBI NIH HHS/United States
- 13POST17290043/AHA/American Heart Association-American Stroke Association/United States
- UM1 HL098179/HL/NHLBI NIH HHS/United States
- U01 HL131003/HL/NHLBI NIH HHS/United States
- R01 HL137755/HL/NHLBI NIH HHS/United States
- T32 HL007731/HL/NHLBI NIH HHS/United States
- C06 RR018928/RR/NCRR NIH HHS/United States
- UM1 HL172717/HL/NHLBI NIH HHS/United States
- P01 HL089707/HL/NHLBI NIH HHS/United States
- U01 HL098179/HL/NHLBI NIH HHS/United States
- R01 HL160665/HL/NHLBI NIH HHS/United States
- P01 HL146366/HL/NHLBI NIH HHS/United States
- 861914/AHA/American Heart Association-American Stroke Association/United States
- R01 HL162925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
