

Endoplasmic Reticulum Stress-Associated Neuronal Death and Innate Immune Response in Neurological Diseases
- PMID: 35082783
- PMCID: PMC8784382
- DOI: 10.3389/fimmu.2021.794580
Endoplasmic Reticulum Stress-Associated Neuronal Death and Innate Immune Response in Neurological Diseases
Abstract
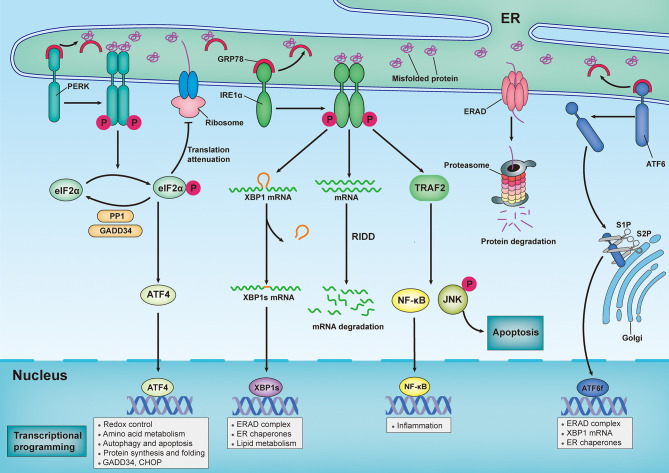
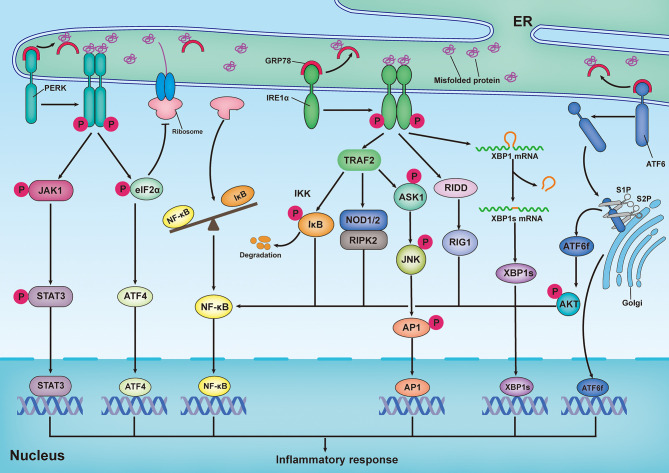
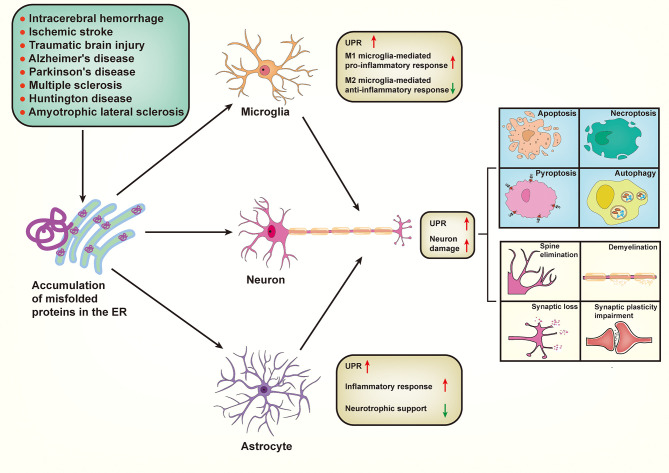
Neuronal death and inflammatory response are two common pathological hallmarks of acute central nervous system injury and chronic degenerative disorders, both of which are closely related to cognitive and motor dysfunction associated with various neurological diseases. Neurological diseases are highly heterogeneous; however, they share a common pathogenesis, that is, the aberrant accumulation of misfolded/unfolded proteins within the endoplasmic reticulum (ER). Fortunately, the cell has intrinsic quality control mechanisms to maintain the proteostasis network, such as chaperone-mediated folding and ER-associated degradation. However, when these control mechanisms fail, misfolded/unfolded proteins accumulate in the ER lumen and contribute to ER stress. ER stress has been implicated in nearly all neurological diseases. ER stress initiates the unfolded protein response to restore proteostasis, and if the damage is irreversible, it elicits intracellular cascades of death and inflammation. With the growing appreciation of a functional association between ER stress and neurological diseases and with the improved understanding of the multiple underlying molecular mechanisms, pharmacological and genetic targeting of ER stress are beginning to emerge as therapeutic approaches for neurological diseases.
Keywords: endoplasmic reticulum stress; inflammatory response; neurological diseases; neuronal death; proteostasis; unfolded protein response.
Copyright © 2022 Shi, Chai, Zhang and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Insights Into the Role of Endoplasmic Reticulum Stress in Infectious Diseases.Front Immunol. 2020 Jan 31;10:3147. doi: 10.3389/fimmu.2019.03147. eCollection 2019. Front Immunol. 2020. PMID: 32082307 Free PMC article. Review.
-
The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities.Front Immunol. 2020 Feb 11;10:3154. doi: 10.3389/fimmu.2019.03154. eCollection 2019. Front Immunol. 2020. PMID: 32117210 Free PMC article. Review.
-
The unfolded protein response: how protein folding became a restrictive aspect for innate immunity and B lymphocytes.Scand J Immunol. 2011 May;73(5):436-48. doi: 10.1111/j.1365-3083.2010.02504.x. Scand J Immunol. 2011. PMID: 21204902 Review.
-
Unfolded Protein Response of the Endoplasmic Reticulum in Tumor Progression and Immunogenicity.Oxid Med Cell Longev. 2017;2017:2969271. doi: 10.1155/2017/2969271. Epub 2017 Dec 21. Oxid Med Cell Longev. 2017. PMID: 29430279 Free PMC article. Review.
-
Protein Quality Control in the Endoplasmic Reticulum and Cancer.Int J Mol Sci. 2018 Oct 3;19(10):3020. doi: 10.3390/ijms19103020. Int J Mol Sci. 2018. PMID: 30282948 Free PMC article. Review.
Cited by
-
Mitophagy-associated programmed neuronal death and neuroinflammation.Front Immunol. 2024 Oct 2;15:1460286. doi: 10.3389/fimmu.2024.1460286. eCollection 2024. Front Immunol. 2024. PMID: 39416788 Free PMC article. Review.
-
Meta-analysis and in-silico functional characterization of the SNCA variant rs356220 in Parkinson's disease.Sci Rep. 2025 Jul 2;15(1):23358. doi: 10.1038/s41598-025-04435-0. Sci Rep. 2025. PMID: 40603942 Free PMC article.
-
Azetidine-2-Carboxylic Acid-Induced Oligodendrogliopathy: Relevance to the Pathogenesis of Multiple Sclerosis.J Neuropathol Exp Neurol. 2022 May 20;81(6):414-433. doi: 10.1093/jnen/nlac028. J Neuropathol Exp Neurol. 2022. PMID: 35521963 Free PMC article.
-
Induction of Unfolded Protein Response by Tannic Acid Triggers Apoptosis in MDA-MB-231 Breast Cancer Cells.Asian Pac J Cancer Prev. 2023 Jun 1;24(6):2029-2035. doi: 10.31557/APJCP.2023.24.6.2029. Asian Pac J Cancer Prev. 2023. PMID: 37378933 Free PMC article.
-
TSG-6 inhibits hypertrophic scar fibroblast proliferation by regulating IRE1α/TRAF2/NF-κB signalling.Int Wound J. 2023 Apr;20(4):1008-1019. doi: 10.1111/iwj.13950. Epub 2022 Sep 2. Int Wound J. 2023. PMID: 36056472 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical