

Aberrant upregulation of the glycolytic enzyme PFKFB3 in CLN7 neuronal ceroid lipofuscinosis
- PMID: 35087090
- PMCID: PMC8795187
- DOI: 10.1038/s41467-022-28191-1
Aberrant upregulation of the glycolytic enzyme PFKFB3 in CLN7 neuronal ceroid lipofuscinosis
Abstract
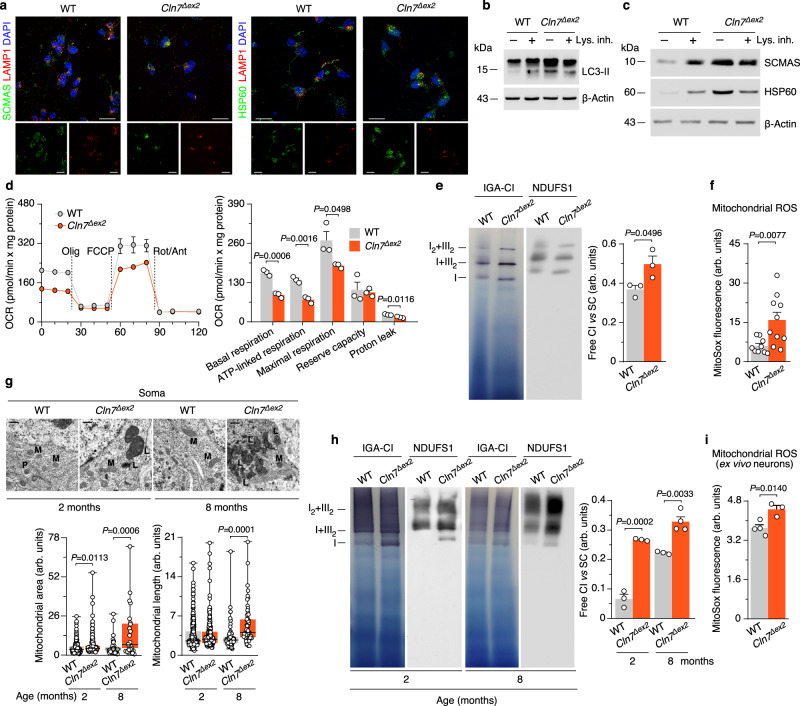
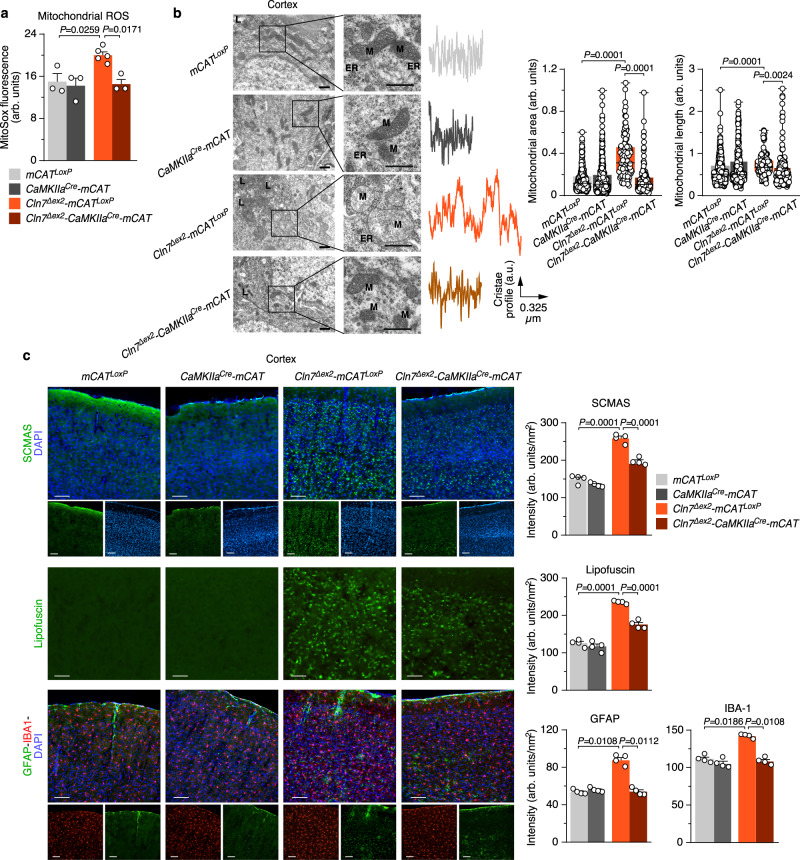
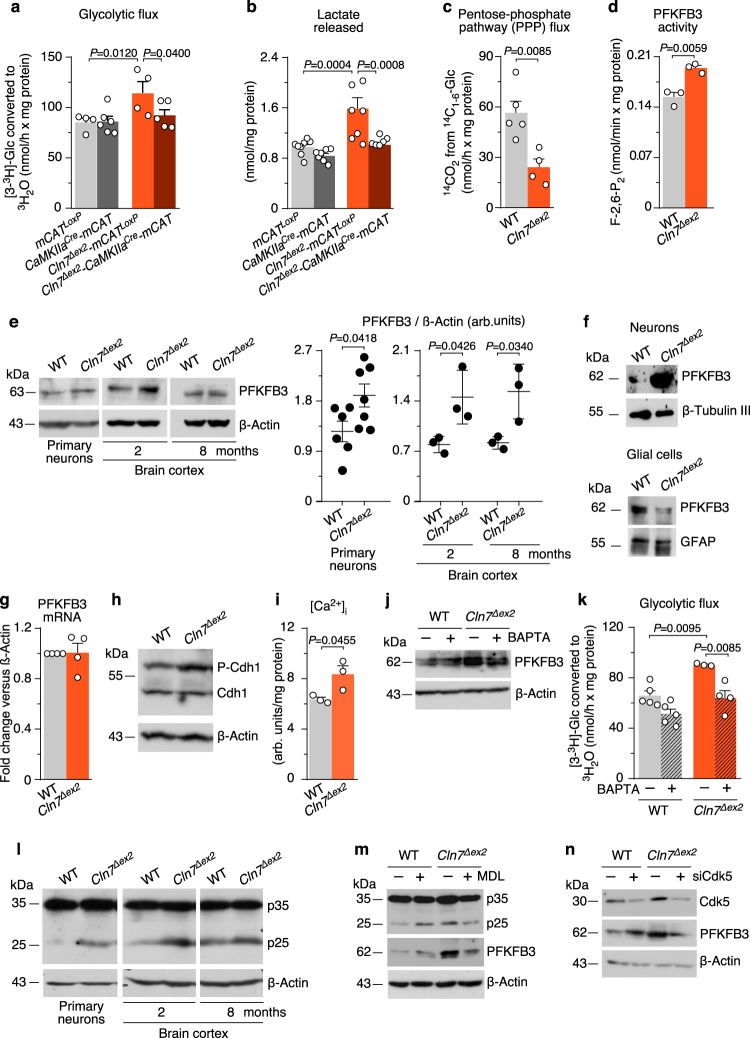
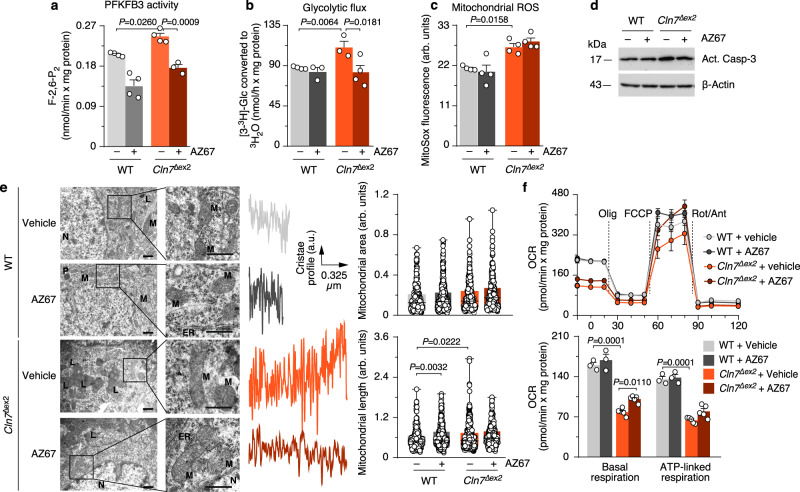
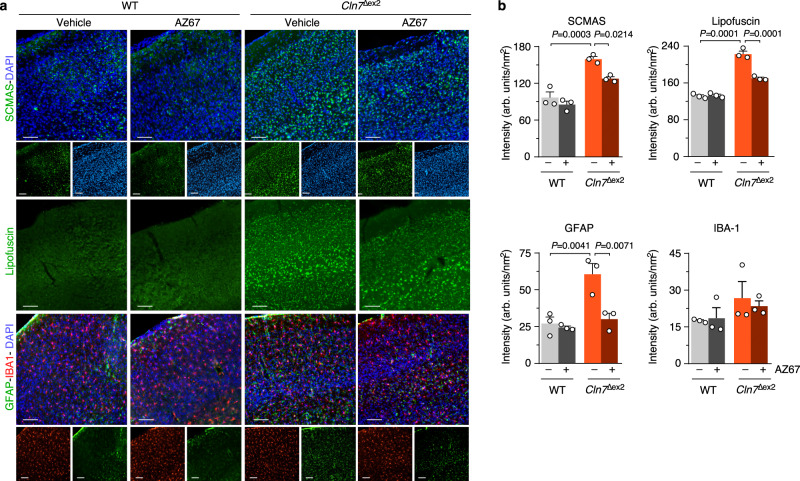
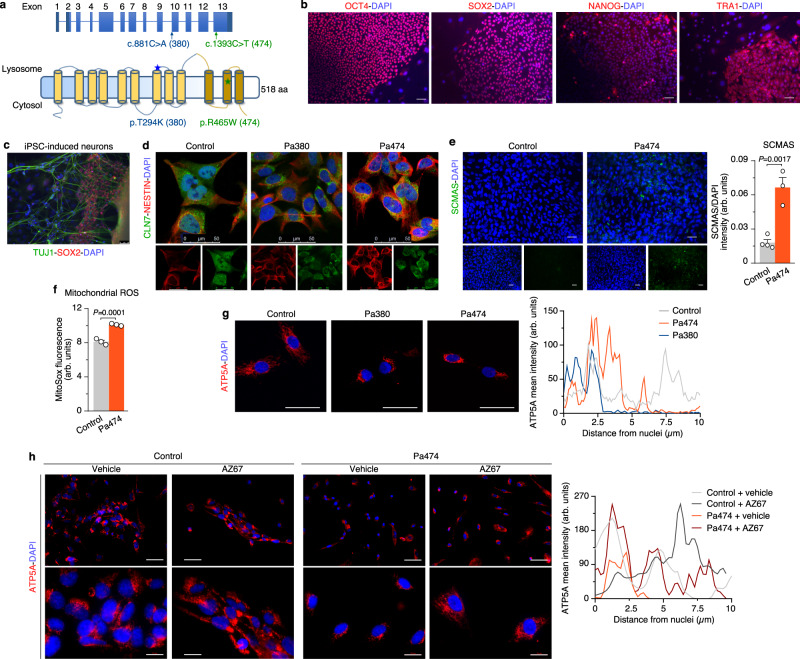
CLN7 neuronal ceroid lipofuscinosis is an inherited lysosomal storage neurodegenerative disease highly prevalent in children. CLN7/MFSD8 gene encodes a lysosomal membrane glycoprotein, but the biochemical processes affected by CLN7-loss of function are unexplored thus preventing development of potential treatments. Here, we found, in the Cln7∆ex2 mouse model of CLN7 disease, that failure in autophagy causes accumulation of structurally and bioenergetically impaired neuronal mitochondria. In vivo genetic approach reveals elevated mitochondrial reactive oxygen species (mROS) in Cln7∆ex2 neurons that mediates glycolytic enzyme PFKFB3 activation and contributes to CLN7 pathogenesis. Mechanistically, mROS sustains a signaling cascade leading to protein stabilization of PFKFB3, normally unstable in healthy neurons. Administration of the highly selective PFKFB3 inhibitor AZ67 in Cln7∆ex2 mouse brain in vivo and in CLN7 patients-derived cells rectifies key disease hallmarks. Thus, aberrant upregulation of the glycolytic enzyme PFKFB3 in neurons may contribute to CLN7 pathogenesis and targeting PFKFB3 could alleviate this and other lysosomal storage diseases.
© 2022. The Author(s).
Conflict of interest statement
P.O.F. is a shareholder and O.B. is an employee of Gero Discovery LLC, a company developing PFKFB3 inhibitors. The remaining authors declare no competing interests.
Figures
References
-
- Mole SE, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 2019;18:107–116. - PubMed
-
- Aiello C, et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2009;30:E530–E540. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
