

Multi-kingdom microbiota analyses identify bacterial-fungal interactions and biomarkers of colorectal cancer across cohorts
- PMID: 35087227
- PMCID: PMC8813618
- DOI: 10.1038/s41564-021-01030-7
Multi-kingdom microbiota analyses identify bacterial-fungal interactions and biomarkers of colorectal cancer across cohorts
Abstract
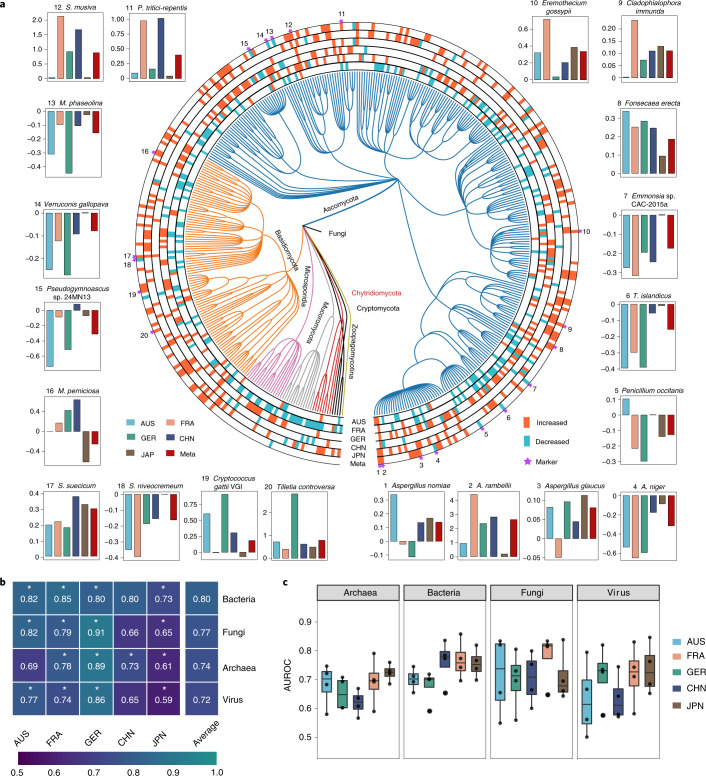
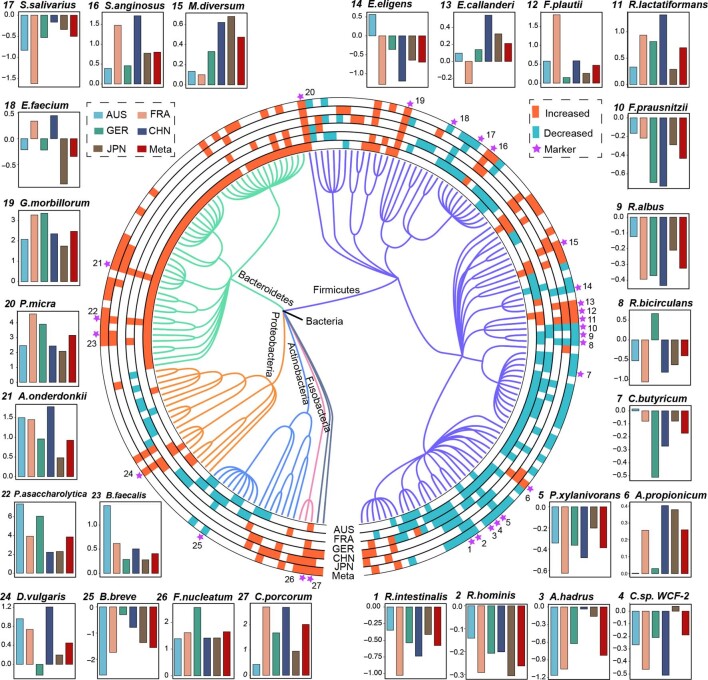
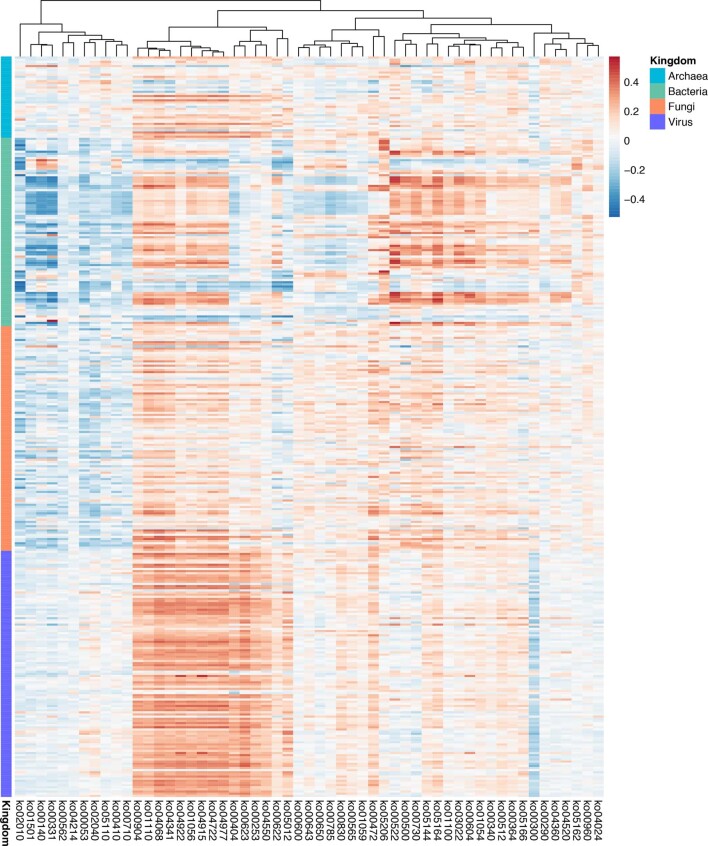
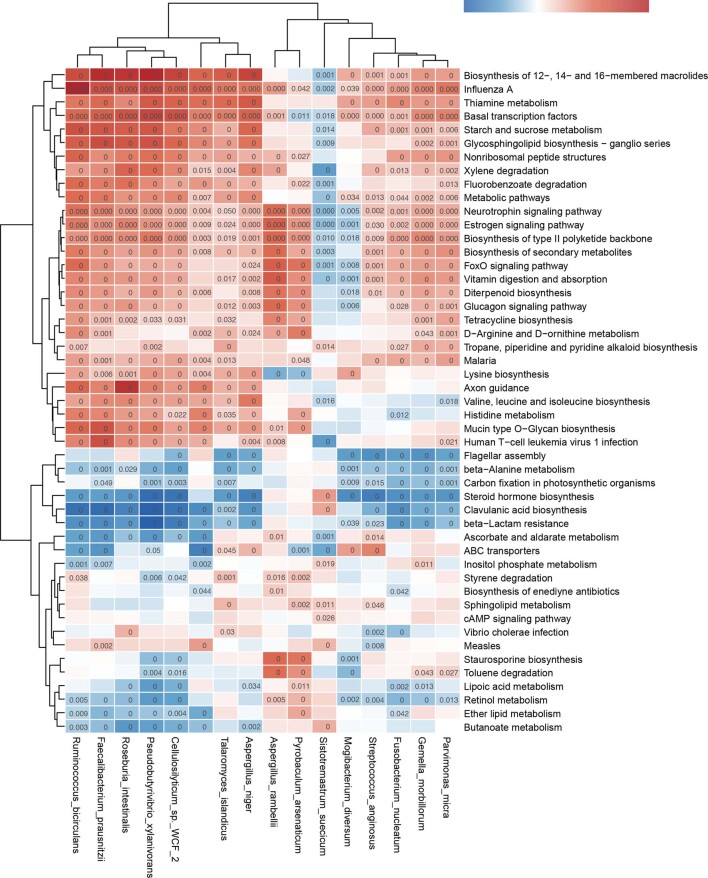
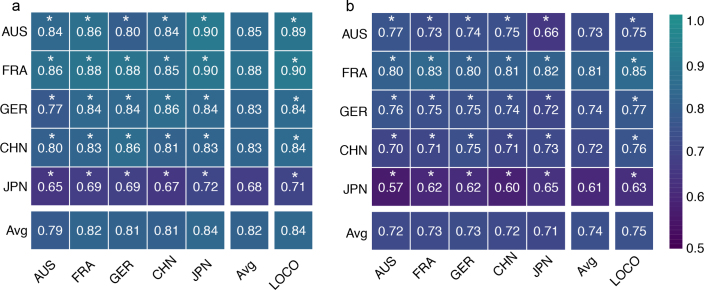
Despite recent progress in our understanding of the association between the gut microbiome and colorectal cancer (CRC), multi-kingdom gut microbiome dysbiosis in CRC across cohorts is unexplored. We investigated four-kingdom microbiota alterations using CRC metagenomic datasets of 1,368 samples from 8 distinct geographical cohorts. Integrated analysis identified 20 archaeal, 27 bacterial, 20 fungal and 21 viral species for each single-kingdom diagnostic model. However, our data revealed superior diagnostic accuracy for models constructed with multi-kingdom markers, in particular the addition of fungal species. Specifically, 16 multi-kingdom markers including 11 bacterial, 4 fungal and 1 archaeal feature, achieved good performance in diagnosing patients with CRC (area under the receiver operating characteristic curve (AUROC) = 0.83) and maintained accuracy across 3 independent cohorts. Coabundance analysis of the ecological network revealed associations between bacterial and fungal species, such as Talaromyces islandicus and Clostridium saccharobutylicum. Using metagenome shotgun sequencing data, the predictive power of the microbial functional potential was explored and elevated D-amino acid metabolism and butanoate metabolism were observed in CRC. Interestingly, the diagnostic model based on functional EggNOG genes achieved high accuracy (AUROC = 0.86). Collectively, our findings uncovered CRC-associated microbiota common across cohorts and demonstrate the applicability of multi-kingdom and functional markers as CRC diagnostic tools and, potentially, as therapeutic targets for the treatment of CRC.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures










References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
