

Multi-omics analyses of the ulcerative colitis gut microbiome link Bacteroides vulgatus proteases with disease severity
- PMID: 35087228
- PMCID: PMC8852248
- DOI: 10.1038/s41564-021-01050-3
Multi-omics analyses of the ulcerative colitis gut microbiome link Bacteroides vulgatus proteases with disease severity
Abstract
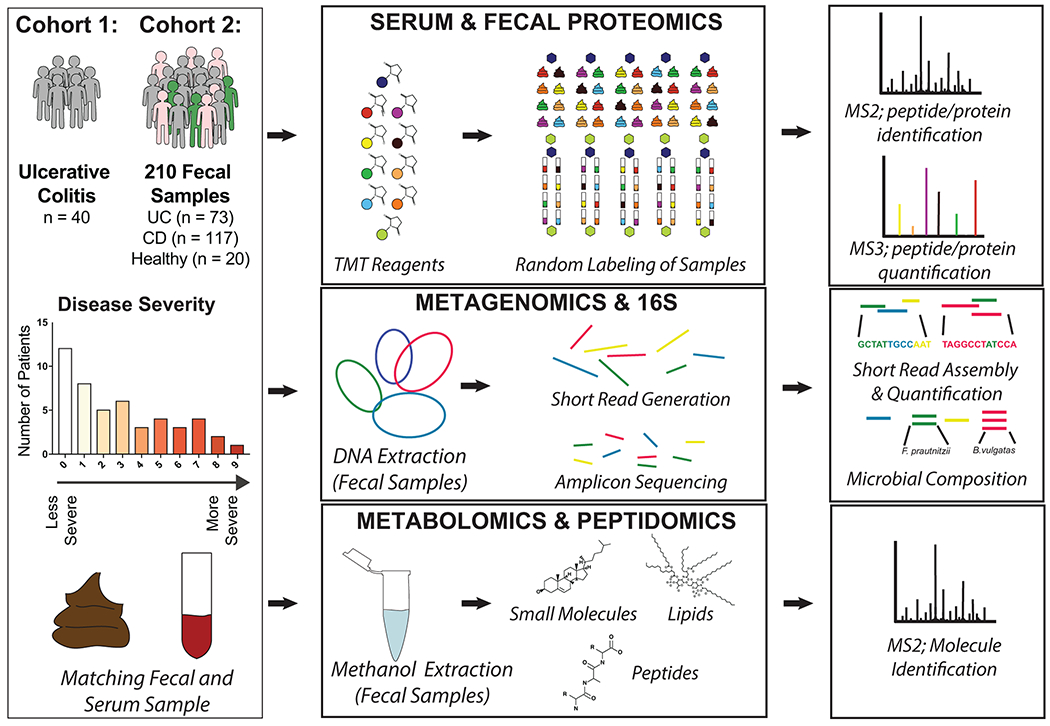
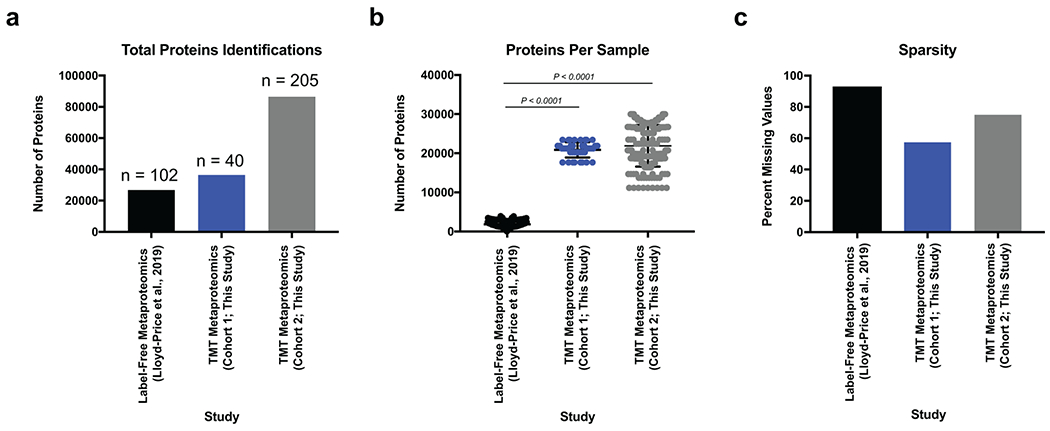
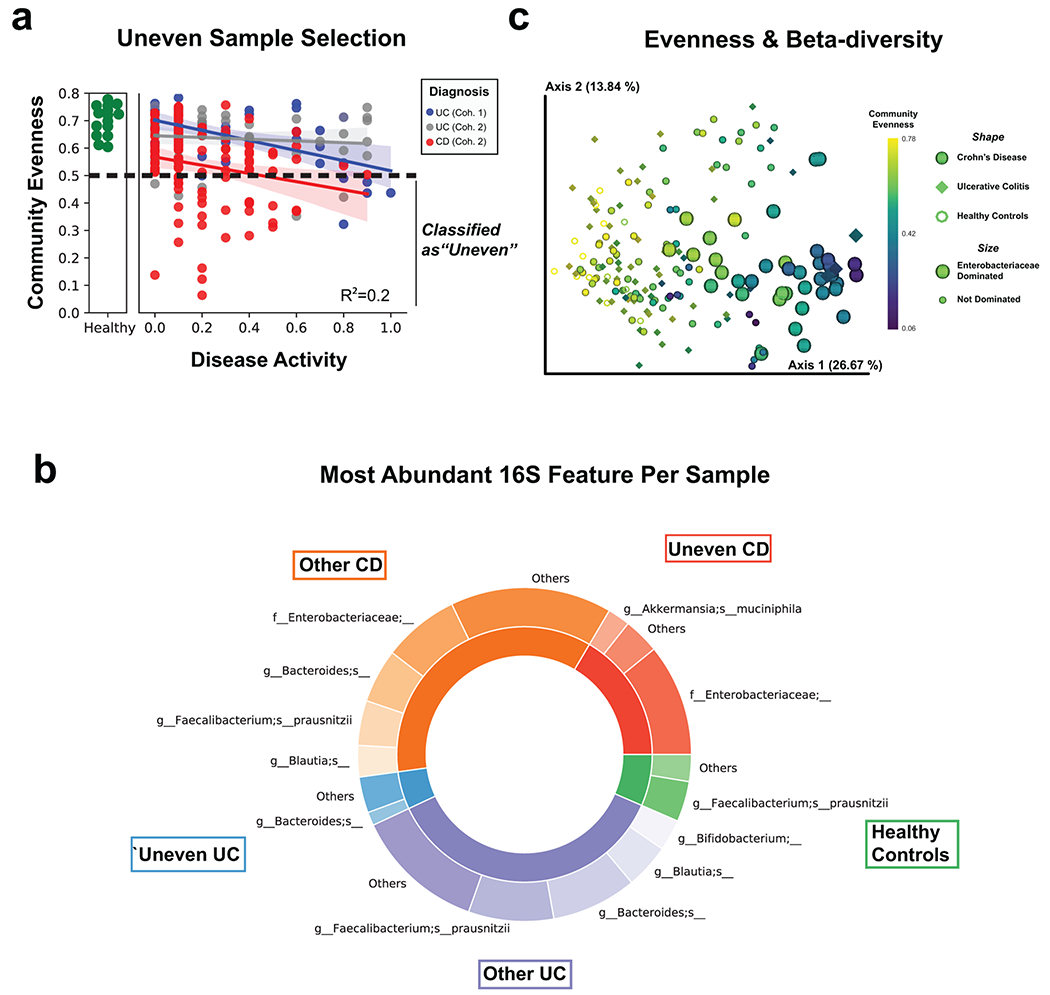
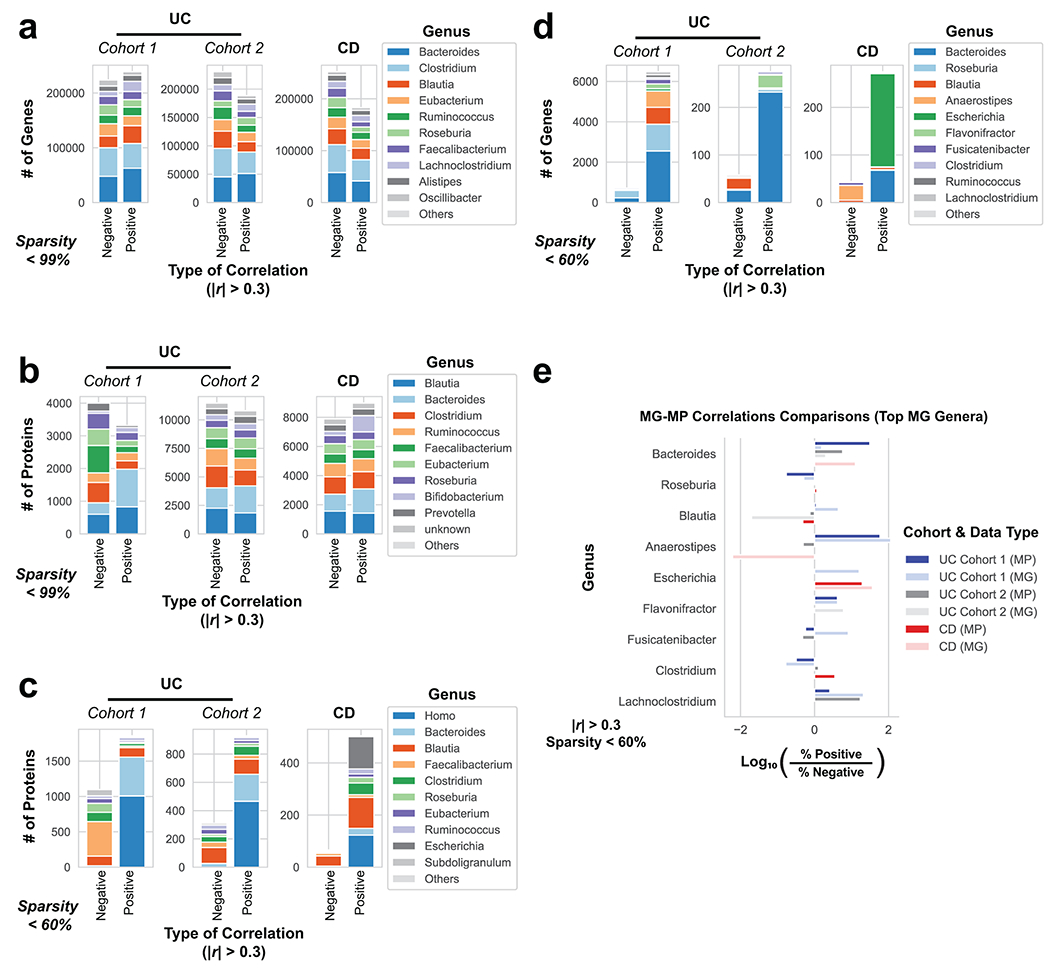
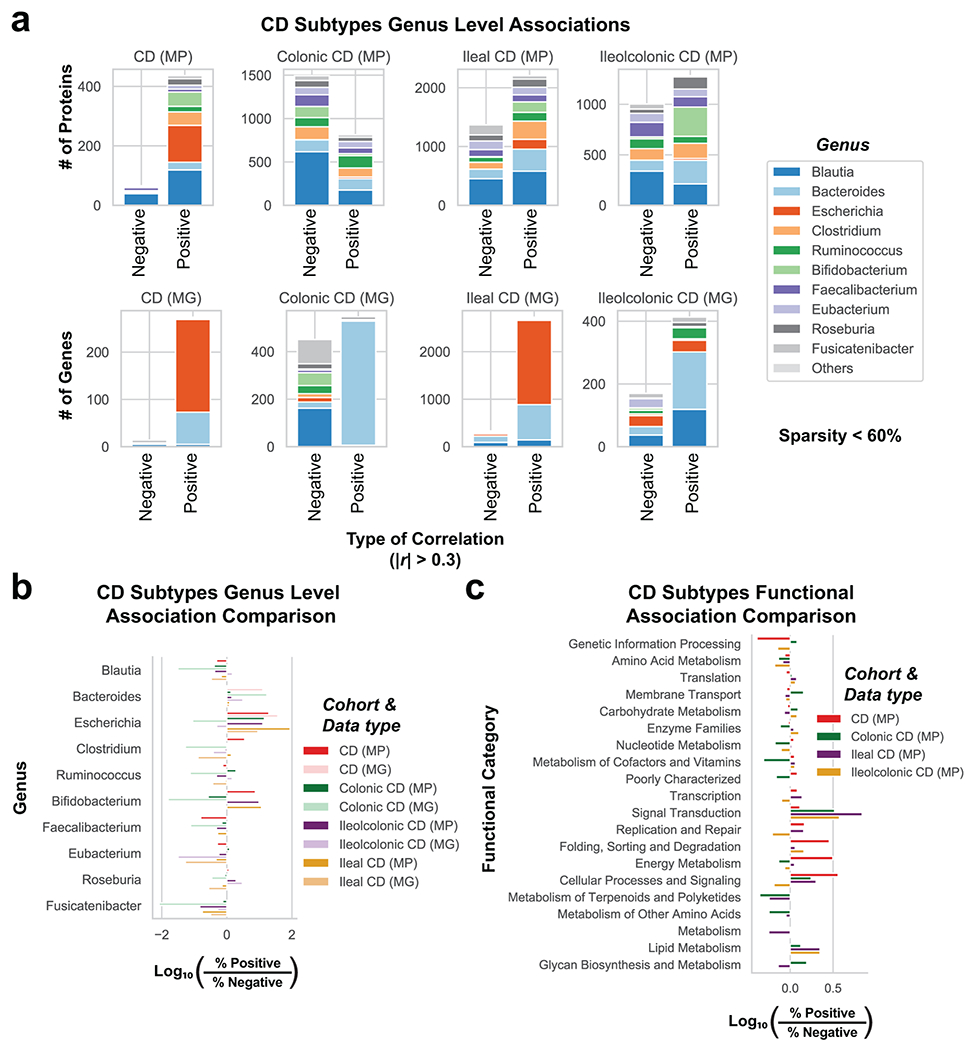
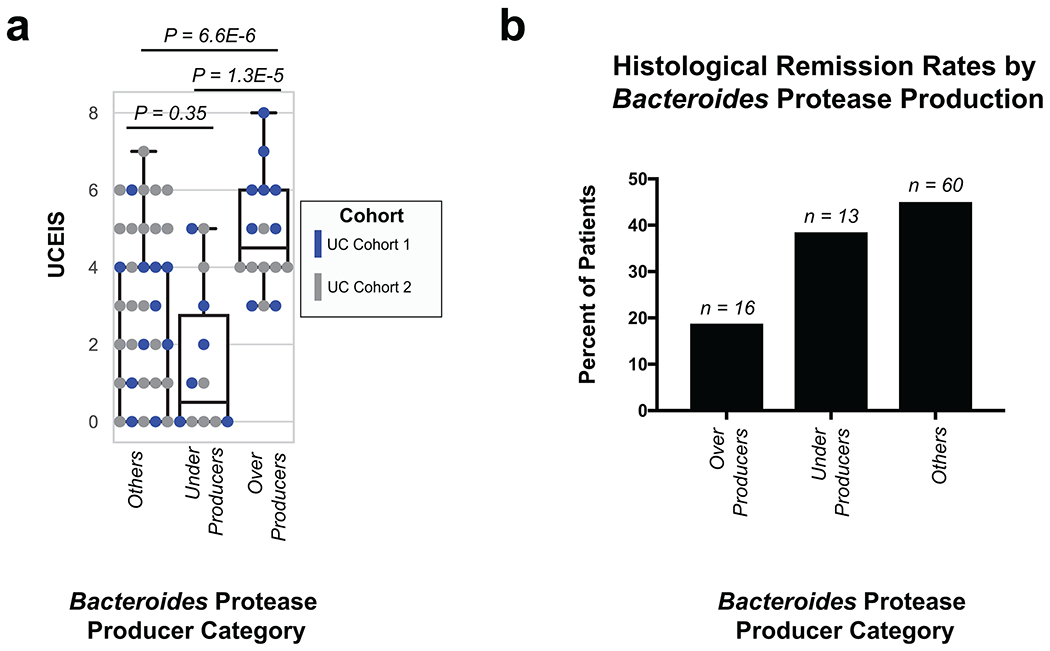
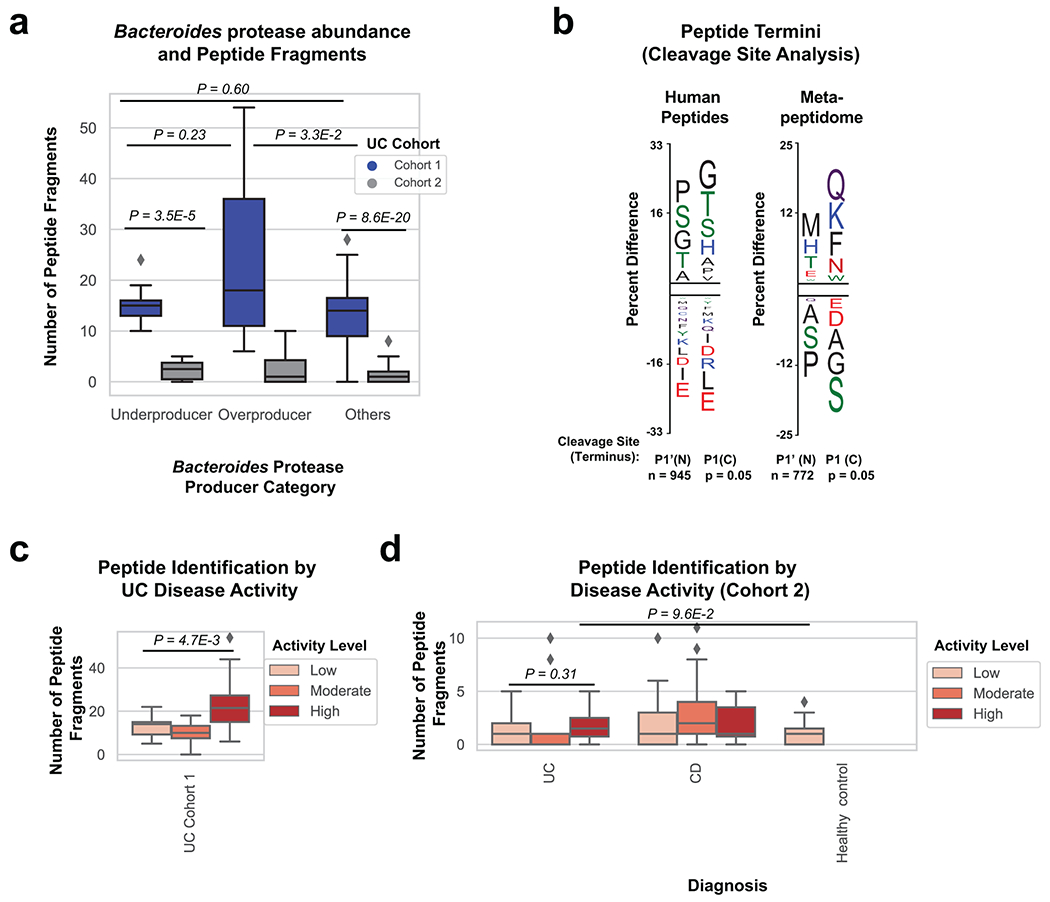
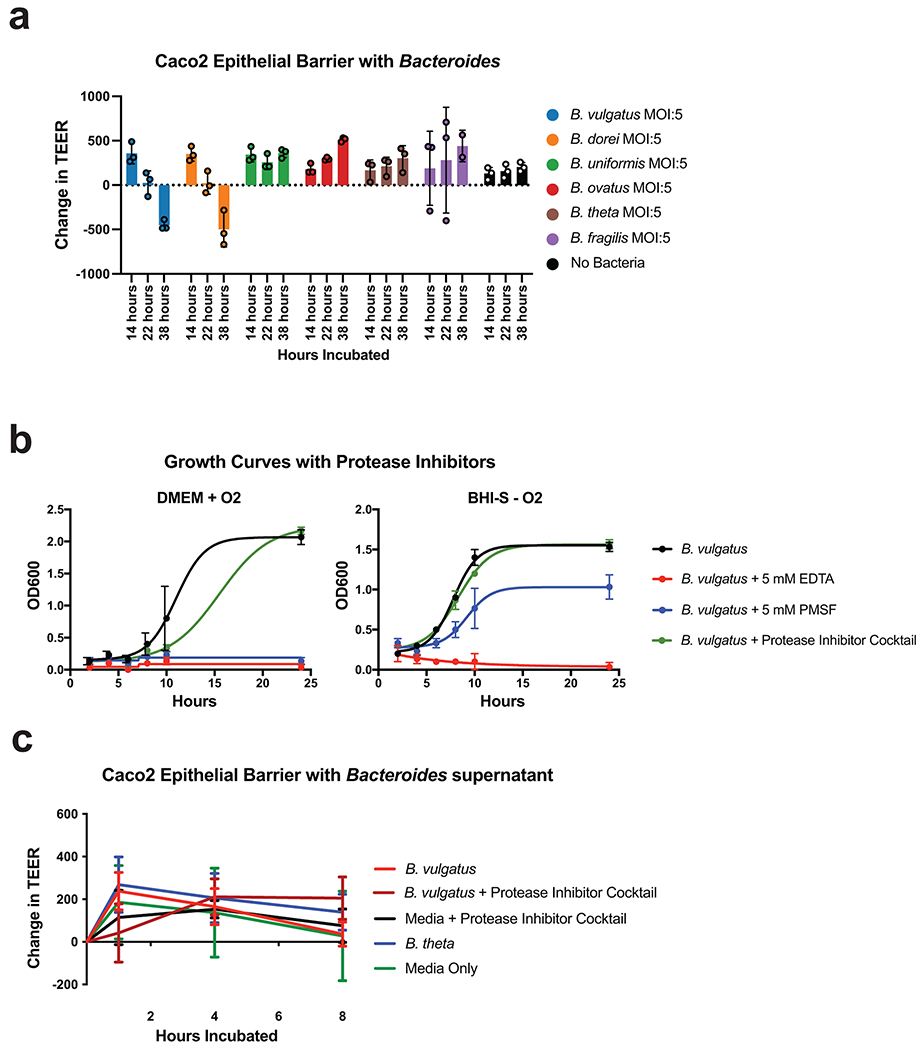
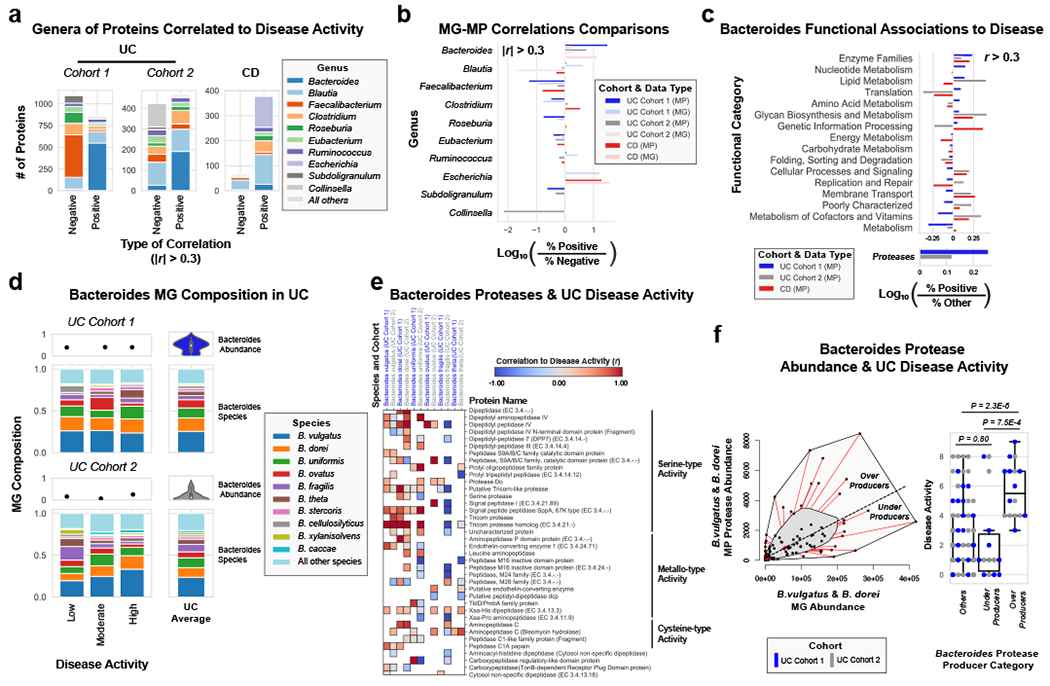
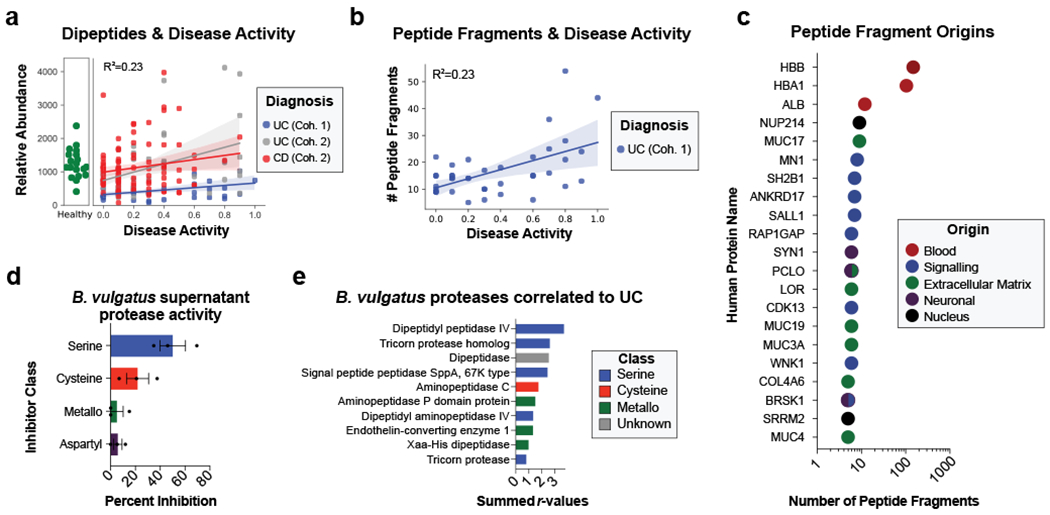
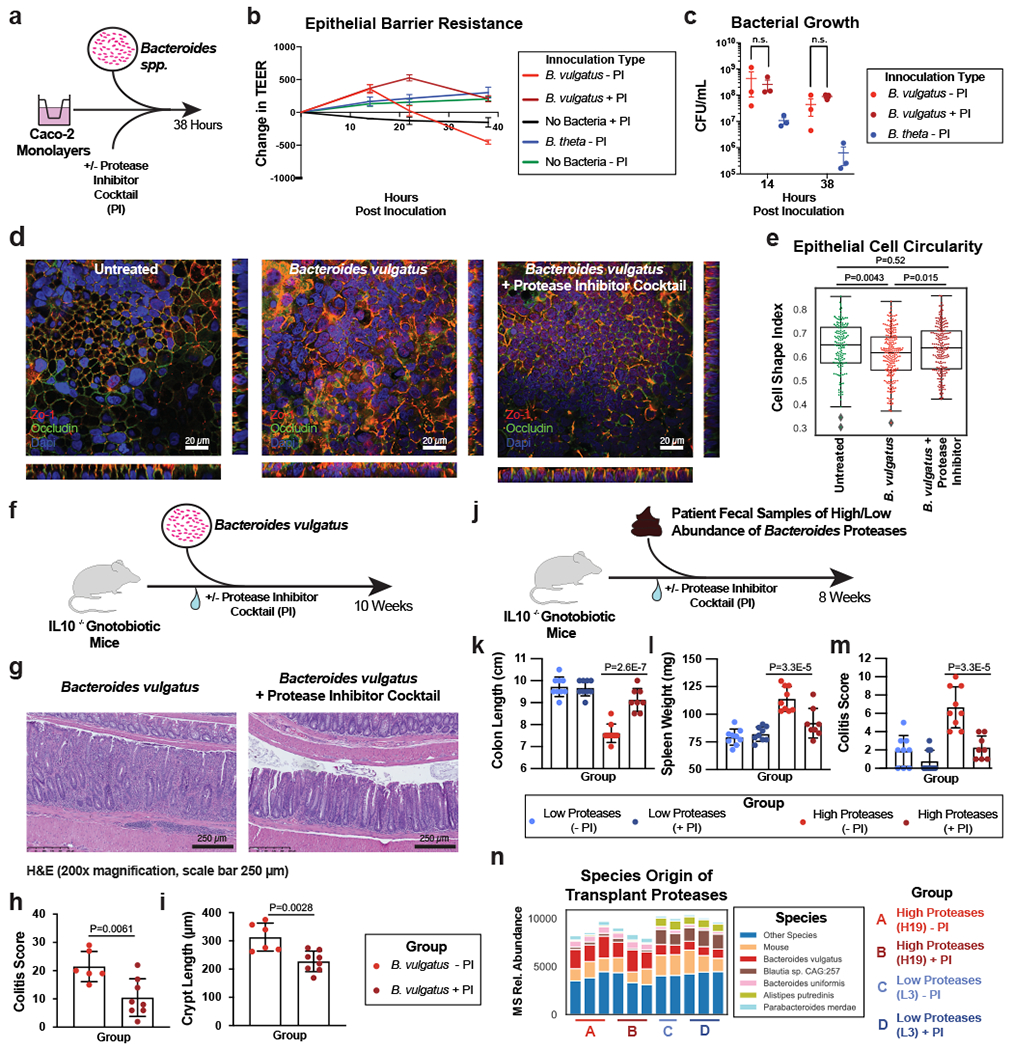
Ulcerative colitis (UC) is driven by disruptions in host-microbiota homoeostasis, but current treatments exclusively target host inflammatory pathways. To understand how host-microbiota interactions become disrupted in UC, we collected and analysed six faecal- or serum-based omic datasets (metaproteomic, metabolomic, metagenomic, metapeptidomic and amplicon sequencing profiles of faecal samples and proteomic profiles of serum samples) from 40 UC patients at a single inflammatory bowel disease centre, as well as various clinical, endoscopic and histologic measures of disease activity. A validation cohort of 210 samples (73 UC, 117 Crohn's disease, 20 healthy controls) was collected and analysed separately and independently. Data integration across both cohorts showed that a subset of the clinically active UC patients had an overabundance of proteases that originated from the bacterium Bacteroides vulgatus. To test whether B. vulgatus proteases contribute to UC disease activity, we first profiled B. vulgatus proteases found in patients and bacterial cultures. Use of a broad-spectrum protease inhibitor improved B. vulgatus-induced barrier dysfunction in vitro, and prevented colitis in B. vulgatus monocolonized, IL10-deficient mice. Furthermore, transplantation of faeces from UC patients with a high abundance of B. vulgatus proteases into germfree mice induced colitis dependent on protease activity. These results, stemming from a multi-omics approach, improve understanding of functional microbiota alterations that drive UC and provide a resource for identifying other pathways that could be inhibited as a strategy to treat this disease.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Code Availability
The code used in analysis and visualization of data is available at
Statistics & Reproducibility
Multi -omic data was collected and analyzed in two independent experiments to increase the likelihood of reproducibility. Sample sizes from each cohort were largely driven by technical and financial constraints as opposed to power analysis, but our sample sizes are similar to those reported in previous publications,,. Several samples were removed after the identification of bacterial blooms or red blood cell contamination as indicated in the metadata. Experiments were randomized during sample processing.
COMPETING INTERESTS
R.H.M., P.S.D and D.J.G. have jointly filed for a patent based on this work (International Application No. PCT/US2020/057784). Over the course of the publication process, R.H.M. started employment at Precidiag Inc., a company which has licensed the patent based on this work. All other authors declare no competing interests.
Figures
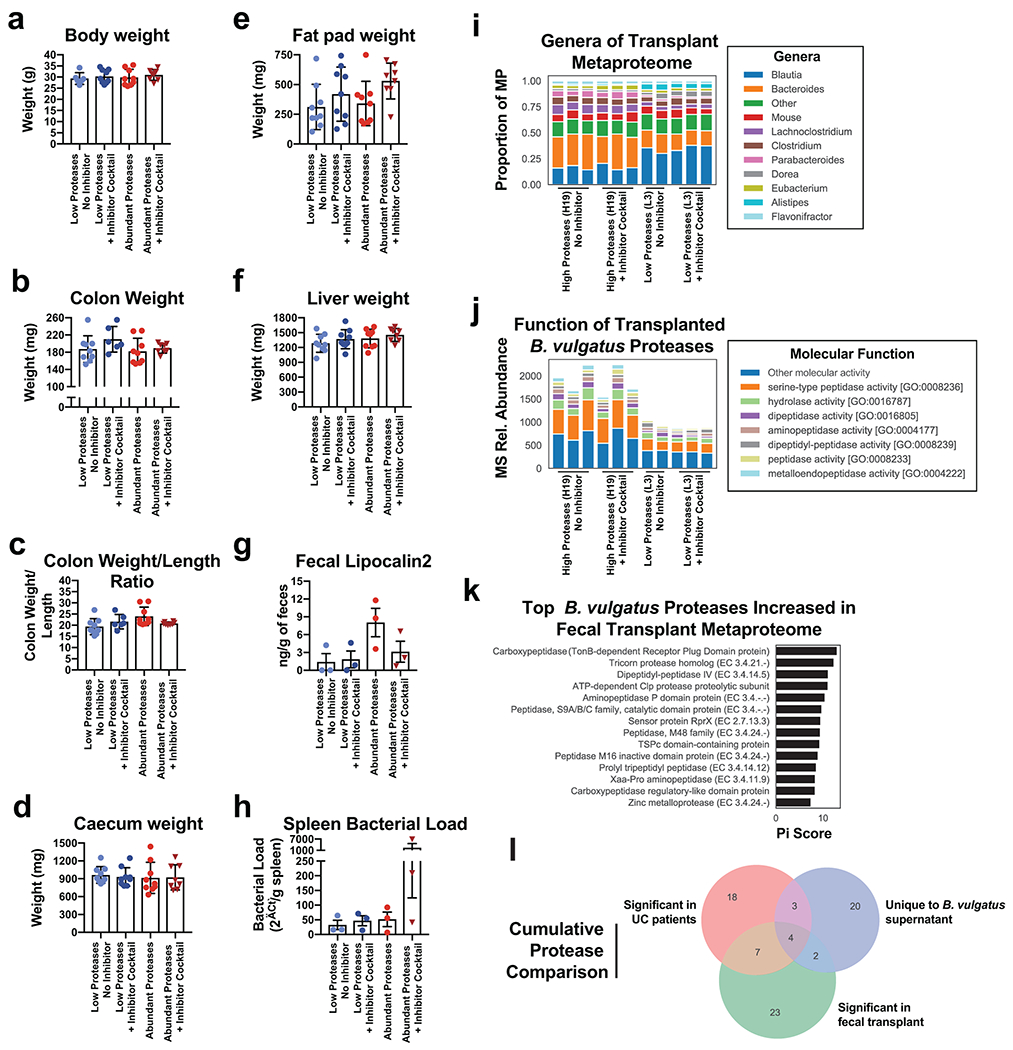
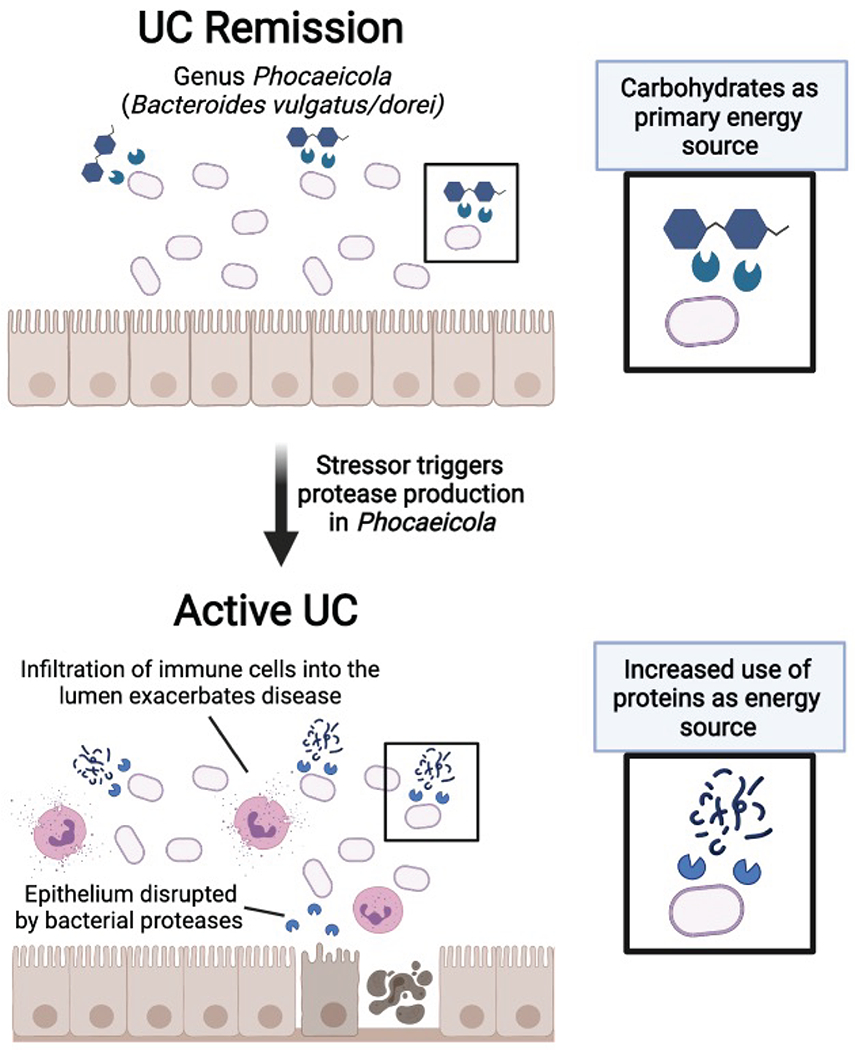
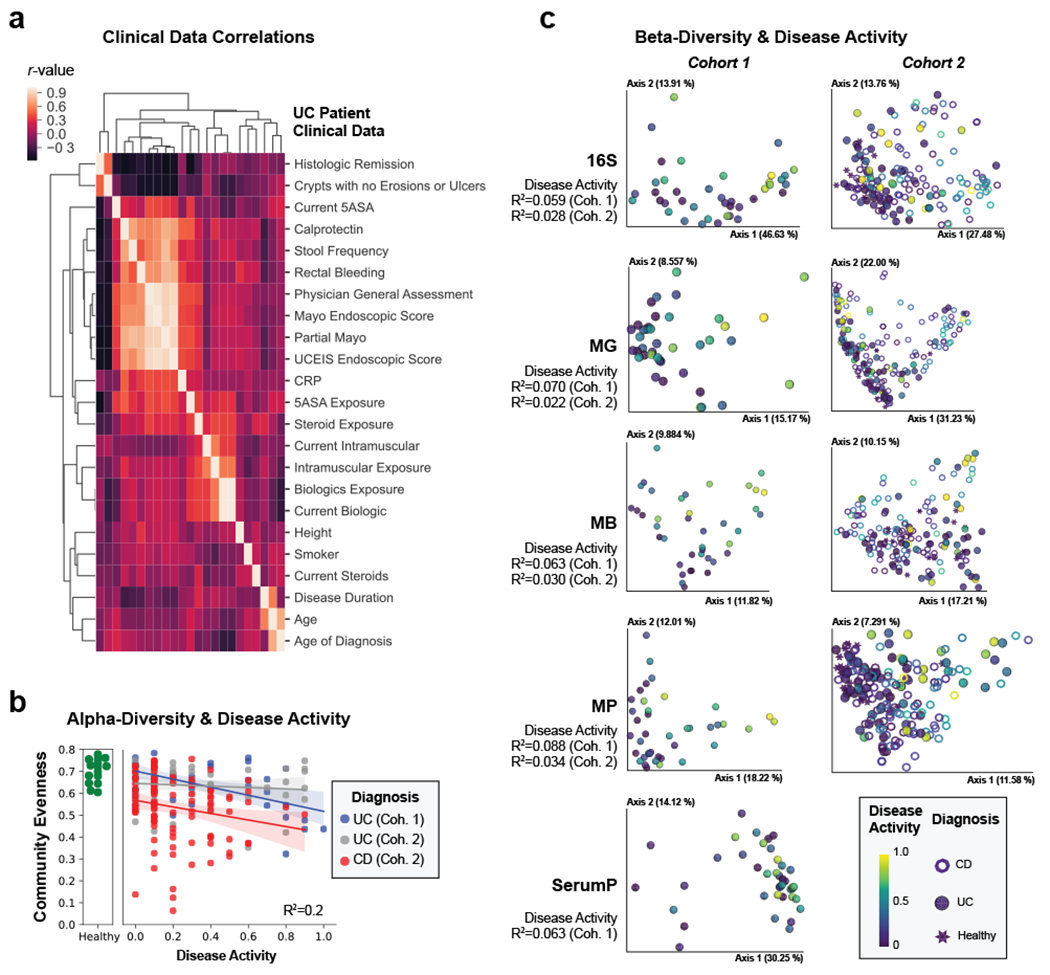
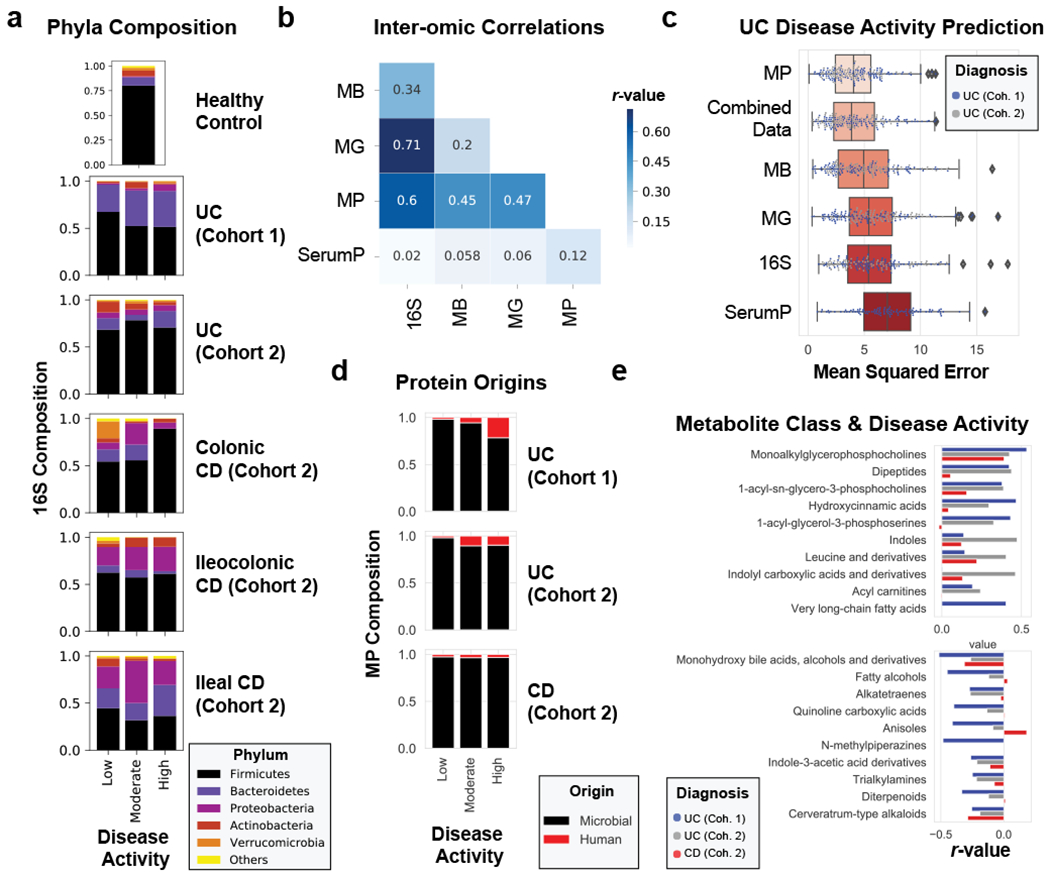
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
