

Multi-omic landscaping of human midbrains identifies disease-relevant molecular targets and pathways in advanced-stage Parkinson's disease
- PMID: 35090094
- PMCID: PMC8797064
- DOI: 10.1002/ctm2.692
Multi-omic landscaping of human midbrains identifies disease-relevant molecular targets and pathways in advanced-stage Parkinson's disease
Abstract
Background: Parkinson's disease (PD) is the second most common neurodegenerative disorder whose prevalence is rapidly increasing worldwide. The molecular mechanisms underpinning the pathophysiology of sporadic PD remain incompletely understood. Therefore, causative therapies are still elusive. To obtain a more integrative view of disease-mediated alterations, we investigated the molecular landscape of PD in human post-mortem midbrains, a region that is highly affected during the disease process.
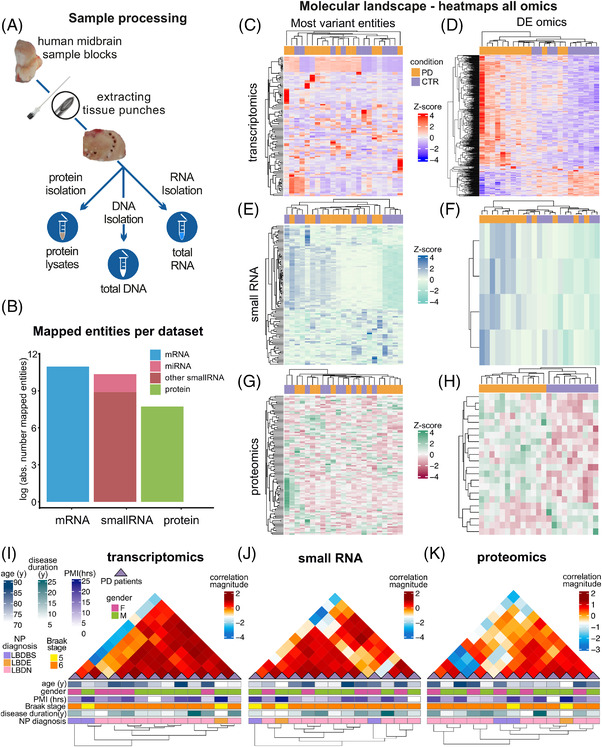
Methods: Tissue from 19 PD patients and 12 controls were obtained from the Parkinson's UK Brain Bank and subjected to multi-omic analyses: small and total RNA sequencing was performed on an Illumina's HiSeq4000, while proteomics experiments were performed in a hybrid triple quadrupole-time of flight mass spectrometer (TripleTOF5600+) following quantitative sequential window acquisition of all theoretical mass spectra. Differential expression analyses were performed with customized frameworks based on DESeq2 (for RNA sequencing) and with Perseus v.1.5.6.0 (for proteomics). Custom pipelines in R were used for integrative studies.
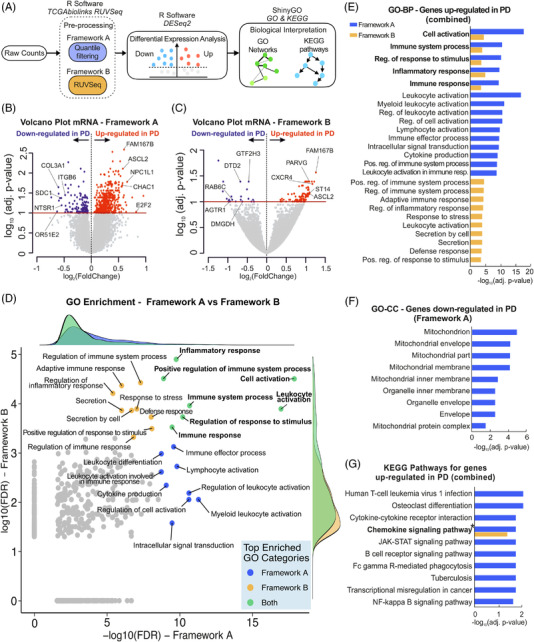
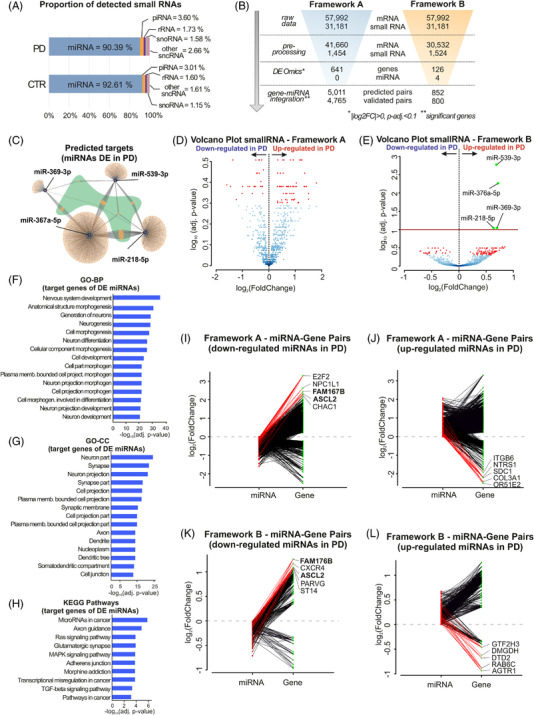
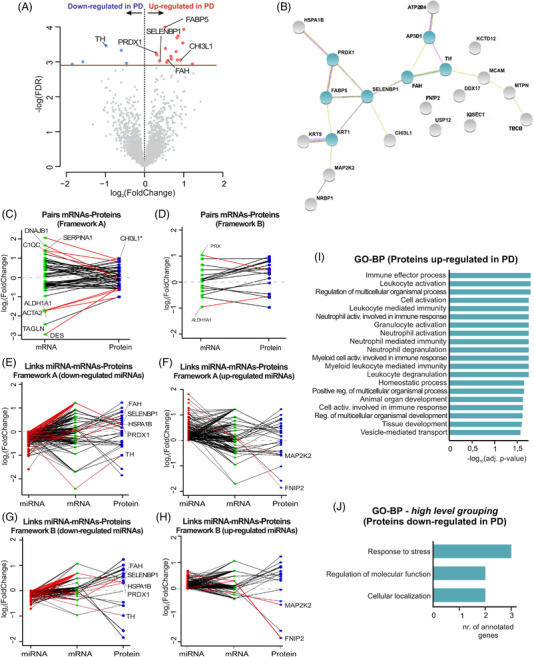
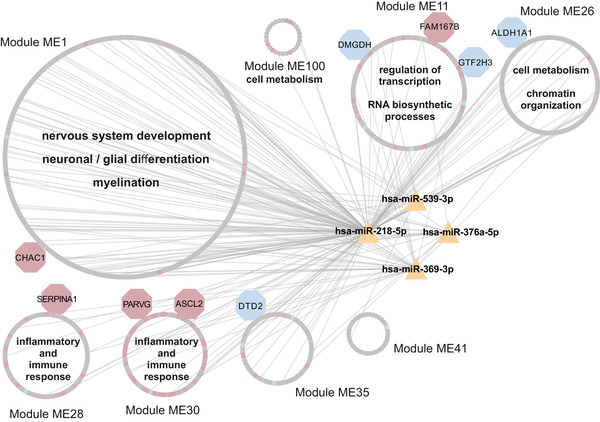
Results: Our analyses revealed multiple deregulated molecular targets linked to known disease mechanisms in PD as well as to novel processes. We have identified and experimentally validated (quantitative real-time polymerase chain reaction/western blotting) several PD-deregulated molecular candidates, including miR-539-3p, miR-376a-5p, miR-218-5p and miR-369-3p, the valid miRNA-mRNA interacting pairs miR-218-5p/RAB6C and miR-369-3p/GTF2H3, as well as multiple proteins, such as CHI3L1, HSPA1B, FNIP2 and TH. Vertical integration of multi-omic analyses allowed validating disease-mediated alterations across different molecular layers. Next to the identification of individual molecular targets in all explored omics layers, functional annotation of differentially expressed molecules showed an enrichment of pathways related to neuroinflammation, mitochondrial dysfunction and defects in synaptic function.
Conclusions: This comprehensive assessment of PD-affected and control human midbrains revealed multiple molecular targets and networks that are relevant to the disease mechanism of advanced PD. The integrative analyses of multiple omics layers underscore the importance of neuroinflammation, immune response activation, mitochondrial and synaptic dysfunction as putative therapeutic targets for advanced PD.
Keywords: Parkinson disease; data integration; miRNAs; multi-omics.
© 2022 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Feigin VL, Abajobir AA, Abate KH, et al. GBD 2015 Neurological Disorders Collaborator Group . Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. The Lancet Neurology. 2017;16(11):877–897. 10.1016/s1474-4422(17)30299-5 - DOI - PMC - PubMed
-
- Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. 2021;397:P2284‐2303. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical