

Human genetic and immunological determinants of critical COVID-19 pneumonia
- PMID: 35090163
- PMCID: PMC8957595
- DOI: 10.1038/s41586-022-04447-0
Human genetic and immunological determinants of critical COVID-19 pneumonia
Abstract
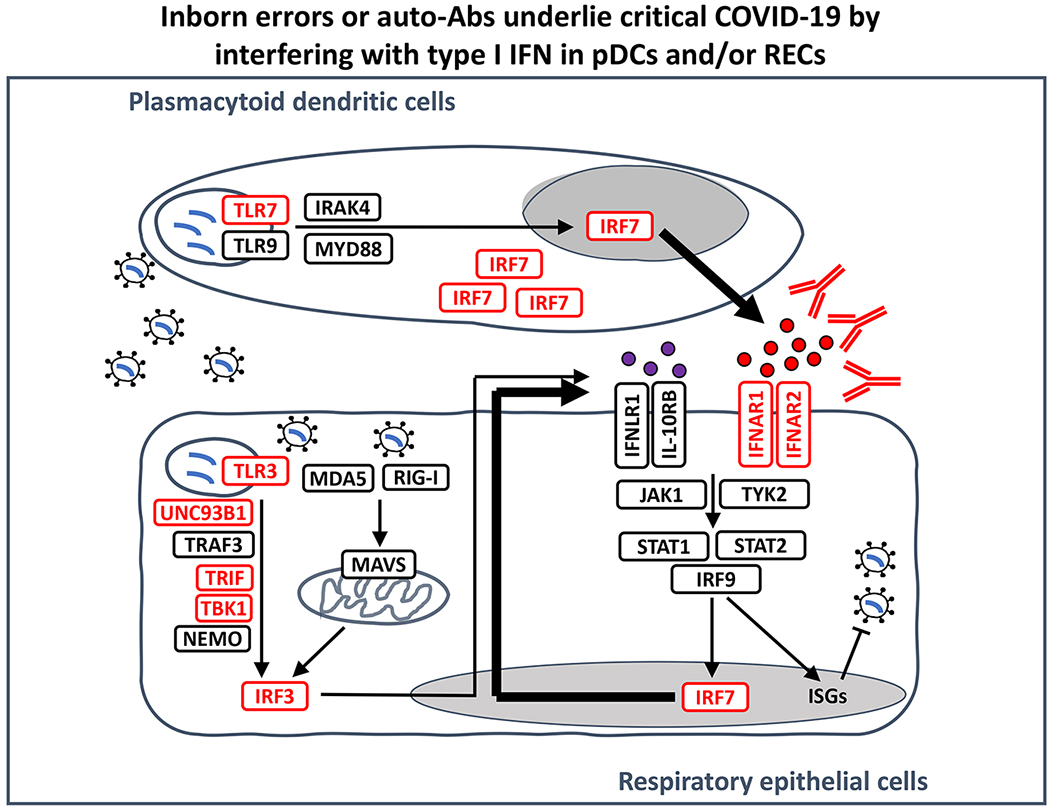
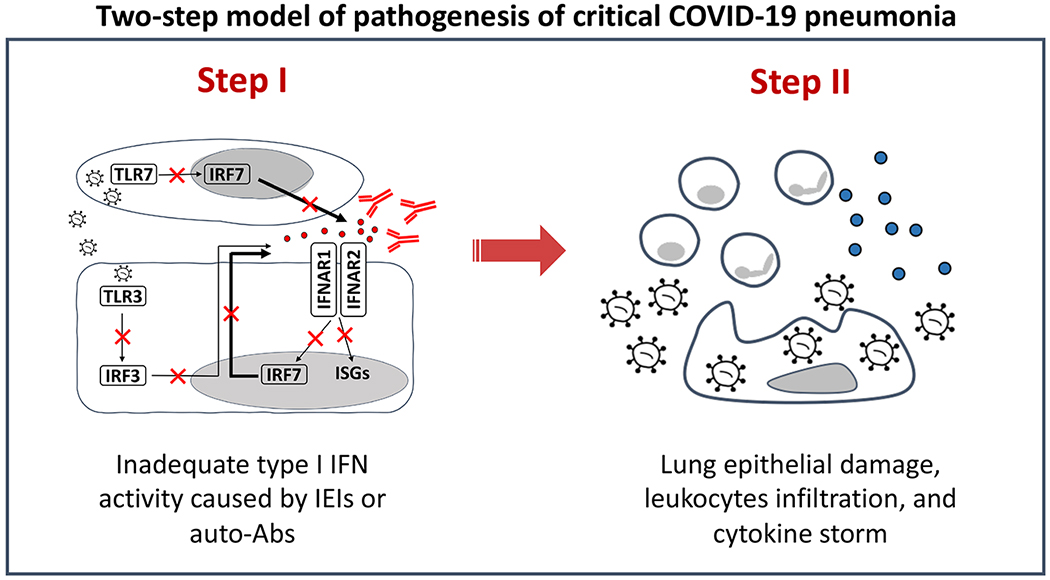
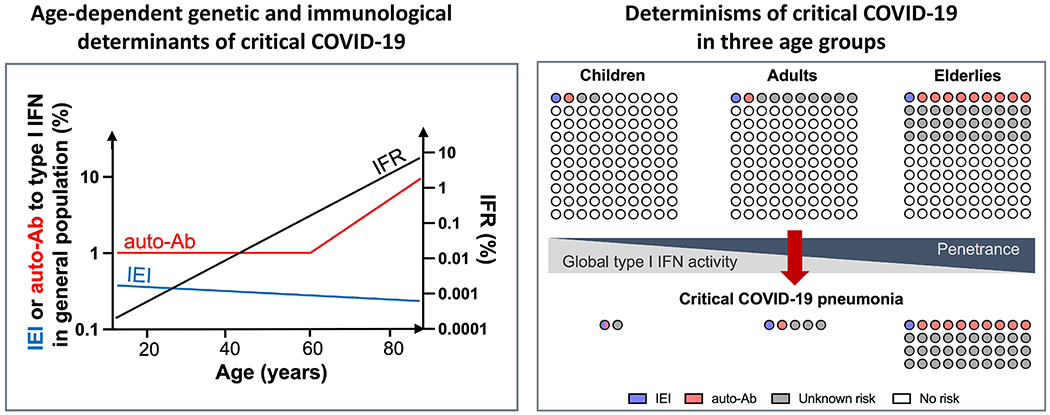
SARS-CoV-2 infection is benign in most individuals but, in around 10% of cases, it triggers hypoxaemic COVID-19 pneumonia, which leads to critical illness in around 3% of cases. The ensuing risk of death (approximately 1% across age and gender) doubles every five years from childhood onwards and is around 1.5 times greater in men than in women. Here we review the molecular and cellular determinants of critical COVID-19 pneumonia. Inborn errors of type I interferons (IFNs), including autosomal TLR3 and X-chromosome-linked TLR7 deficiencies, are found in around 1-5% of patients with critical pneumonia under 60 years old, and a lower proportion in older patients. Pre-existing auto-antibodies neutralizing IFNα, IFNβ and/or IFNω, which are more common in men than in women, are found in approximately 15-20% of patients with critical pneumonia over 70 years old, and a lower proportion in younger patients. Thus, at least 15% of cases of critical COVID-19 pneumonia can be explained. The TLR3- and TLR7-dependent production of type I IFNs by respiratory epithelial cells and plasmacytoid dendritic cells, respectively, is essential for host defence against SARS-CoV-2. In ways that can depend on age and sex, insufficient type I IFN immunity in the respiratory tract during the first few days of infection may account for the spread of the virus, leading to pulmonary and systemic inflammation.
© 2022. Springer Nature Limited.
Conflict of interest statement
Declaration and listing of any financial or non-financial competing interests
The authors declare that there is no financial or non-financial competing interests.
Figures
References
-
- O’Driscoll M, Ribeiro Dos Santos G, Wang L, Cummings DAT, Azman AS, Paireau J, et al. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021; 590(7844):140–145. - PubMed
-
Evidence that the mortality of COVID-19 doubles every 5 years from childhood onward, accounting for a 10,000-fold greater risk at 85 years of age (10%) than at 5 years of age (0.001%).
-
- Sen P, Yamana TK, Kandula S, Galanti M, Shaman J. Burden and characteristics of COVID-19 in the United States during 2020. Nature 2021; 598(7880):338–341. - PubMed
-
- Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (New York, NY) 2020; 370(6515). - PMC - PubMed
-
Report of autosomal inborn errors of type I IFN, including autosomal dominant TLR3, and autosomal recessive IRF7 and IFNAR1 deficiencies, as human genetic and immunological determinants of life-threatening COVID-19 pneumonia.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
