

Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence
- PMID: 35095747
- PMCID: PMC8797140
- DOI: 10.3389/fneur.2021.814174
Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: The Issue of Transgene Persistence
Abstract
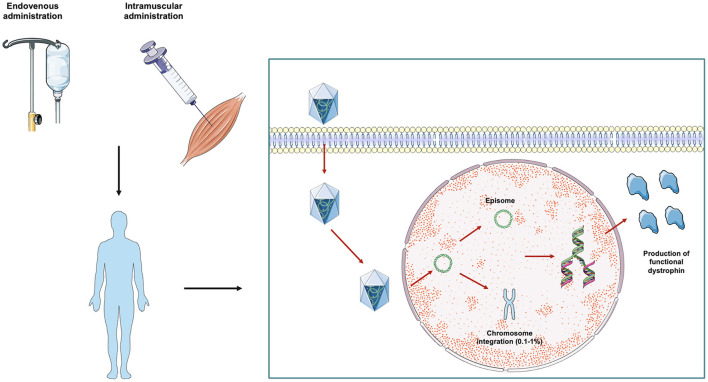
Duchenne muscular dystrophy (DMD) is an X-linked recessive, infancy-onset neuromuscular disorder characterized by progressive muscle weakness and atrophy, leading to delay of motor milestones, loss of autonomous ambulation, respiratory failure, cardiomyopathy, and premature death. DMD originates from mutations in the DMD gene that result in a complete absence of dystrophin. Dystrophin is a cytoskeletal protein which belongs to the dystrophin-associated protein complex, involved in cellular signaling and myofiber membrane stabilization. To date, the few available therapeutic options are aimed at lessening disease progression, but persistent loss of muscle tissue and function and premature death are unavoidable. In this scenario, one of the most promising therapeutic strategies for DMD is represented by adeno-associated virus (AAV)-mediated gene therapy. DMD gene therapy relies on the administration of exogenous micro-dystrophin, a miniature version of the dystrophin gene lacking unnecessary domains and encoding a truncated, but functional, dystrophin protein. Limited transgene persistence represents one of the most significant issues that jeopardize the translatability of DMD gene replacement strategies from the bench to the bedside. Here, we critically review preclinical and clinical studies of AAV-mediated gene therapy in DMD, focusing on long-term transgene persistence in transduced tissues, which can deeply affect effectiveness and sustainability of gene replacement in DMD. We also discuss the role played by the overactivation of the immune host system in limiting long-term expression of genetic material. In this perspective, further studies aimed at better elucidating the need for immune suppression in AAV-treated subjects are warranted in order to allow for life-long therapy in DMD patients.
Keywords: Duchenne muscular dystrophy; adeno-associated virus; dystrophin; gene therapy; microdystrophin; persistence.
Copyright © 2022 Manini, Abati, Nuredini, Corti and Comi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources