

Mechanisms of sex determination and X-chromosome dosage compensation
- PMID: 35100381
- PMCID: PMC8825453
- DOI: 10.1093/genetics/iyab197
Mechanisms of sex determination and X-chromosome dosage compensation
Abstract
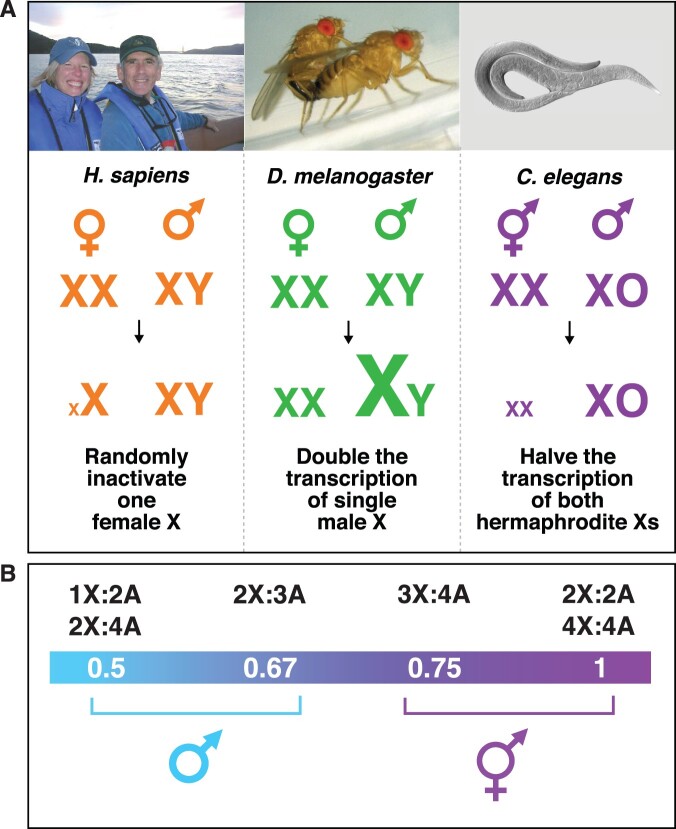
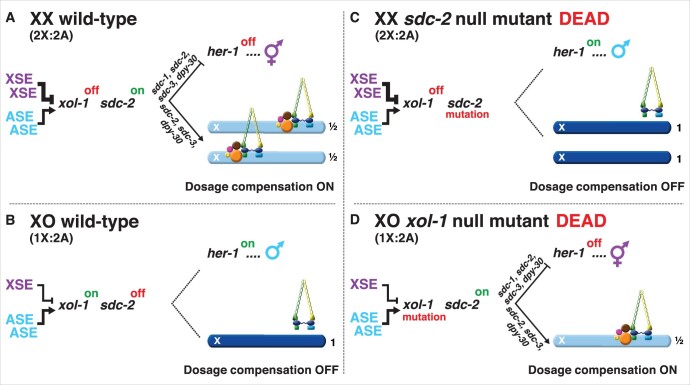
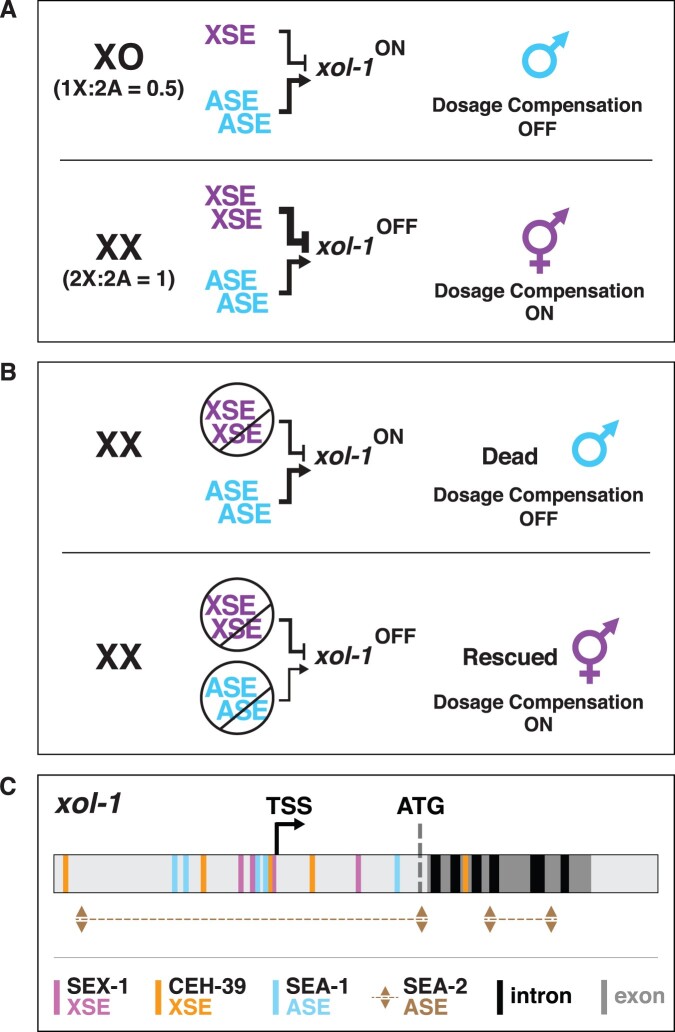
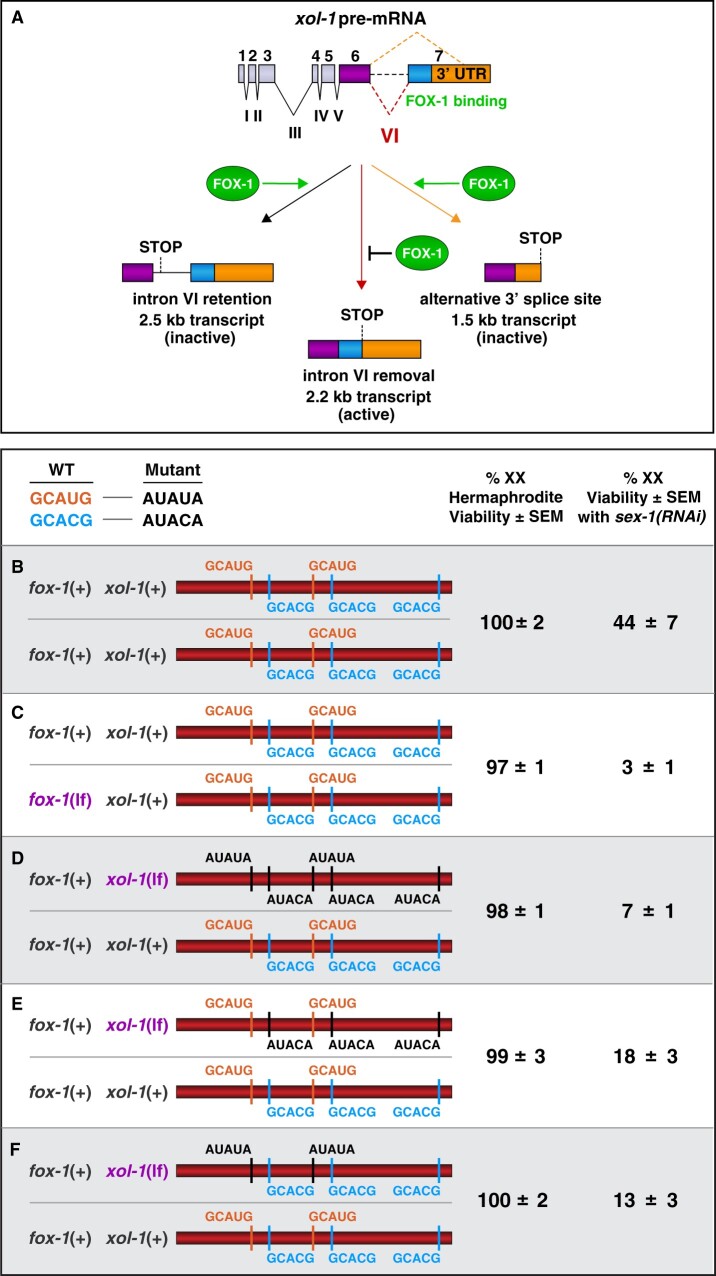
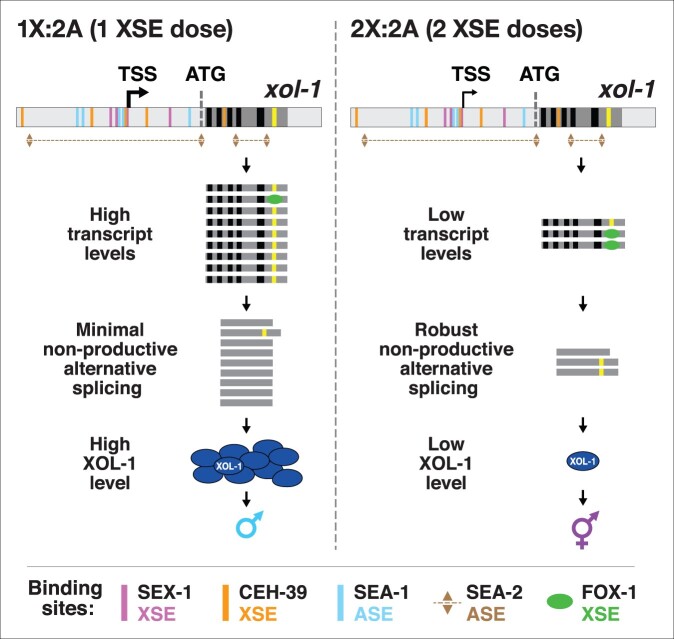
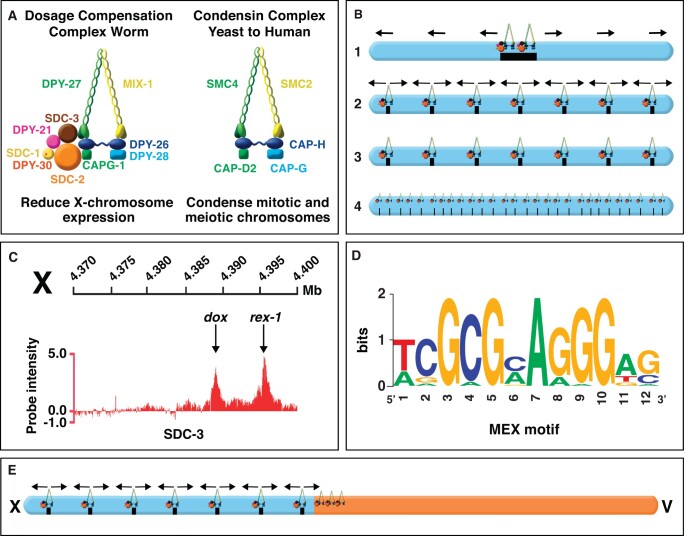
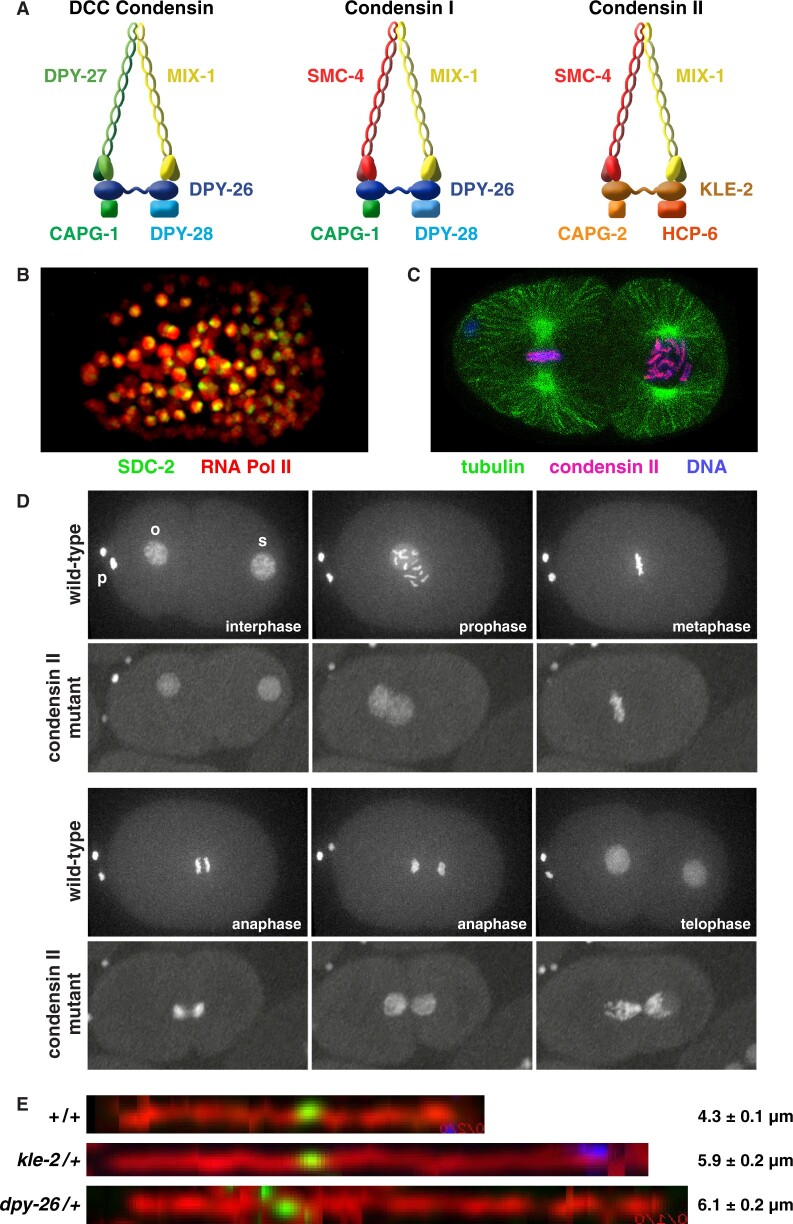
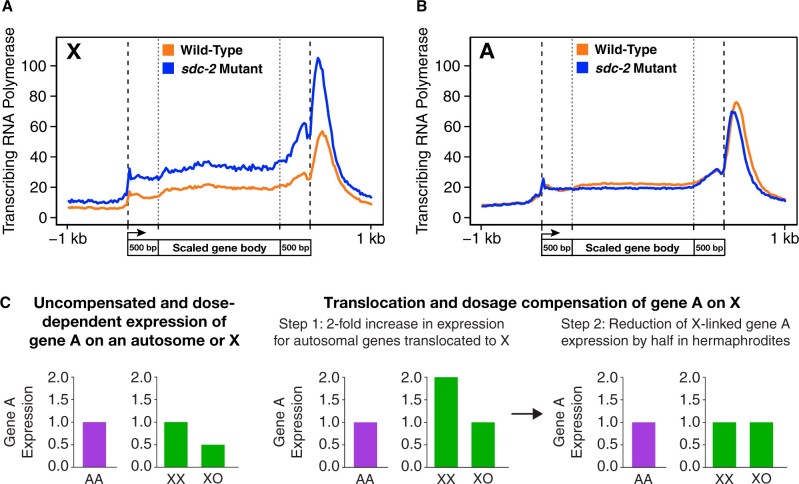
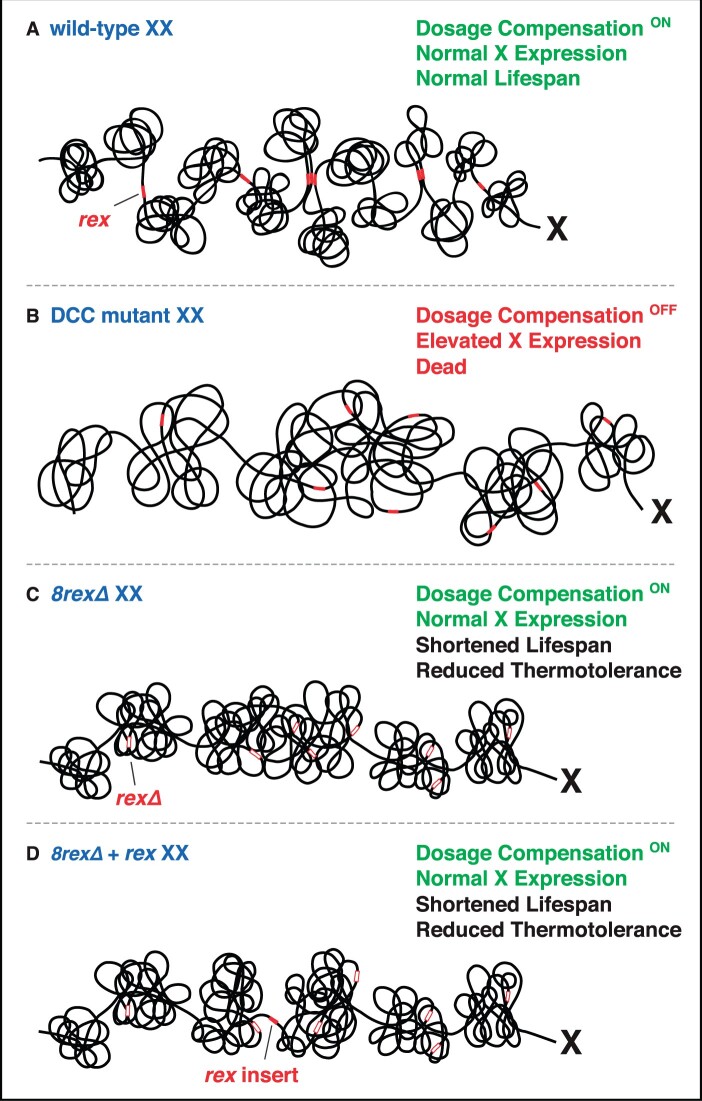
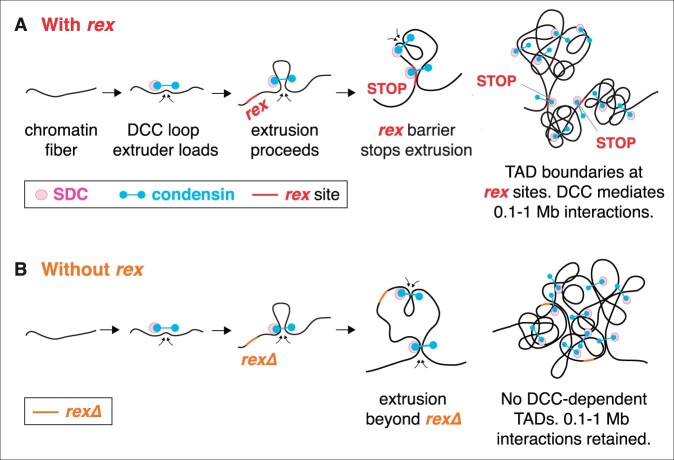
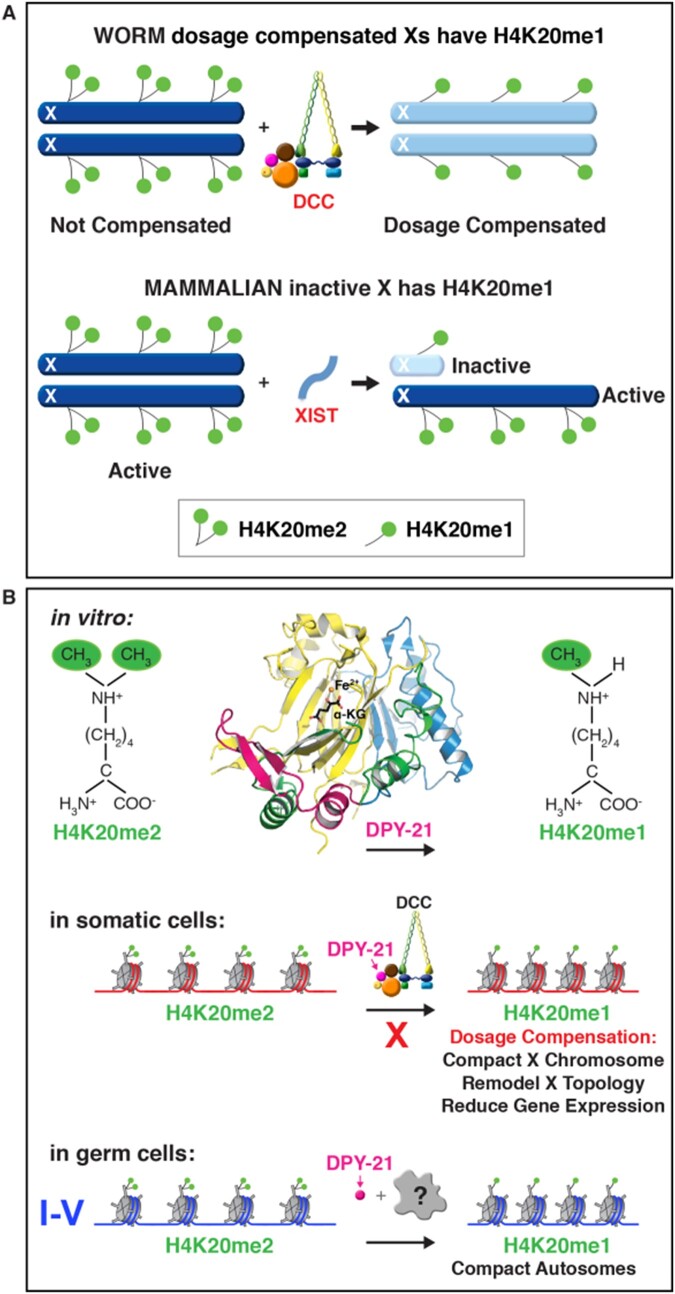
Abnormalities in chromosome number have the potential to disrupt the balance of gene expression and thereby decrease organismal fitness and viability. Such abnormalities occur in most solid tumors and also cause severe developmental defects and spontaneous abortions. In contrast to the imbalances in chromosome dose that cause pathologies, the difference in X-chromosome dose used to determine sexual fate across diverse species is well tolerated. Dosage compensation mechanisms have evolved in such species to balance X-chromosome gene expression between the sexes, allowing them to tolerate the difference in X-chromosome dose. This review analyzes the chromosome counting mechanism that tallies X-chromosome number to determine sex (XO male and XX hermaphrodite) in the nematode Caenorhabditis elegans and the associated dosage compensation mechanism that balances X-chromosome gene expression between the sexes. Dissecting the molecular mechanisms underlying X-chromosome counting has revealed how small quantitative differences in intracellular signals can be translated into dramatically different fates. Dissecting the process of X-chromosome dosage compensation has revealed the interplay between chromatin modification and chromosome structure in regulating gene expression over vast chromosomal territories.
Keywords: Caenorhabditis elegans; WormBook; X-chromosome counting; X-chromosome dosage compensation; cell fate determination; chromosome segregation; chromosome structure; histone modification; mRNA splicing regulation; sex determination; transcriptional regulation.
© The Author(s) 2022. Published by Oxford University Press on behalf of Genetics Society of America.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
