

AGAMEMNON: an Accurate metaGenomics And MEtatranscriptoMics quaNtificatiON analysis suite
- PMID: 35101114
- PMCID: PMC8802518
- DOI: 10.1186/s13059-022-02610-4
AGAMEMNON: an Accurate metaGenomics And MEtatranscriptoMics quaNtificatiON analysis suite
Abstract
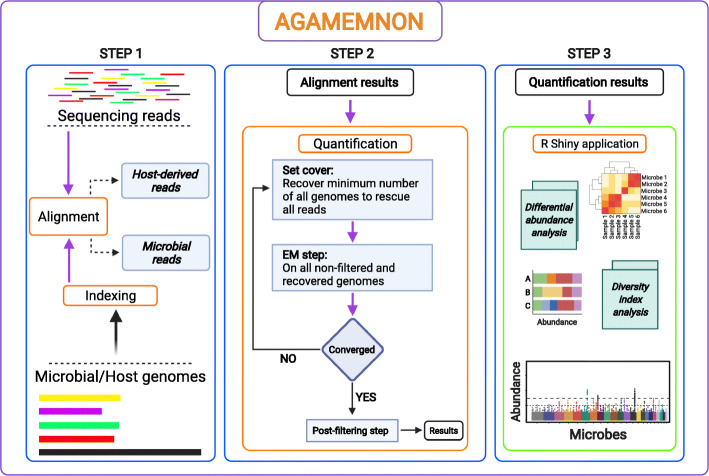
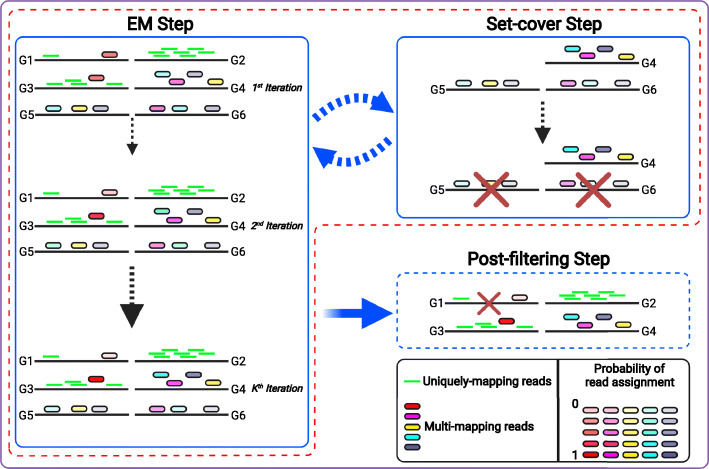
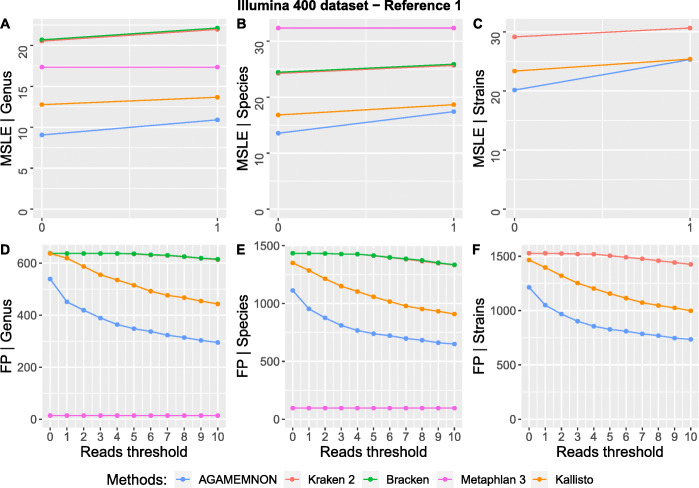
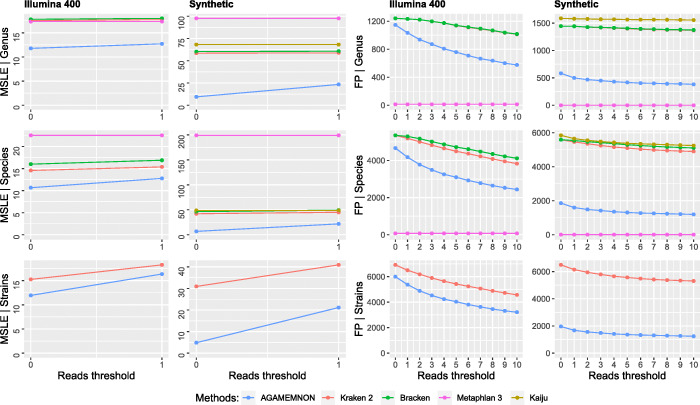
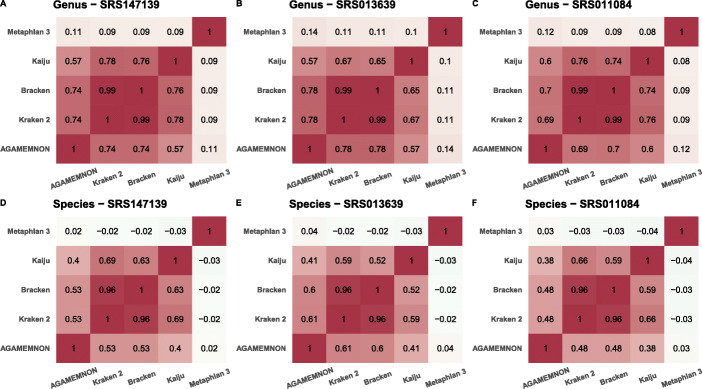
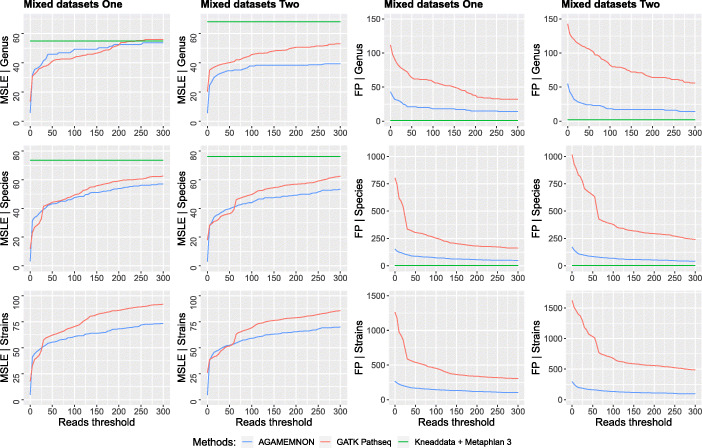
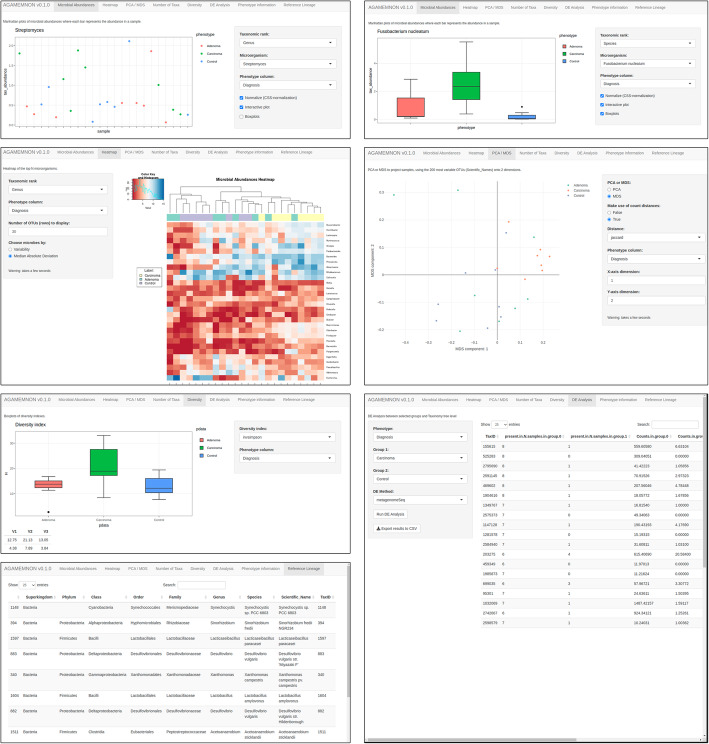
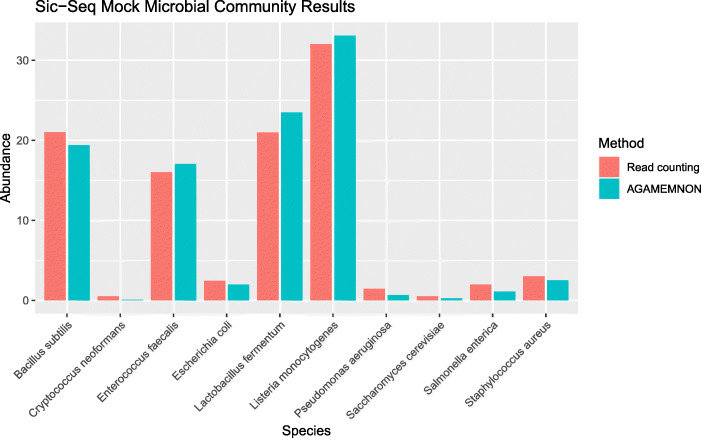
We introduce AGAMEMNON ( https://github.com/ivlachos/agamemnon ) for the acquisition of microbial abundances from shotgun metagenomics and metatranscriptomic samples, single-microbe sequencing experiments, or sequenced host samples. AGAMEMNON delivers accurate abundances at genus, species, and strain resolution. It incorporates a time and space-efficient indexing scheme for fast pattern matching, enabling indexing and analysis of vast datasets with widely available computational resources. Host-specific modules provide exceptional accuracy for microbial abundance quantification from tissue RNA/DNA sequencing, enabling the expansion of experiments lacking metagenomic/metatranscriptomic analyses. AGAMEMNON provides an R-Shiny application, permitting performance of investigations and visualizations from a graphics interface.
Keywords: Computational metagenomics; Identification of contaminants; Microbiome; Quantification of microbial abundances; Time- and space-efficient indexing/alignment.
© 2022. The Author(s).
Conflict of interest statement
RP is a cofounder of Ocean Genomics Inc. The remaining authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
