

Comparison between genetic and pharmaceutical disruption of Ldlr expression for the development of atherosclerosis
- PMID: 35101425
- PMCID: PMC8953673
- DOI: 10.1016/j.jlr.2022.100174
Comparison between genetic and pharmaceutical disruption of Ldlr expression for the development of atherosclerosis
Abstract
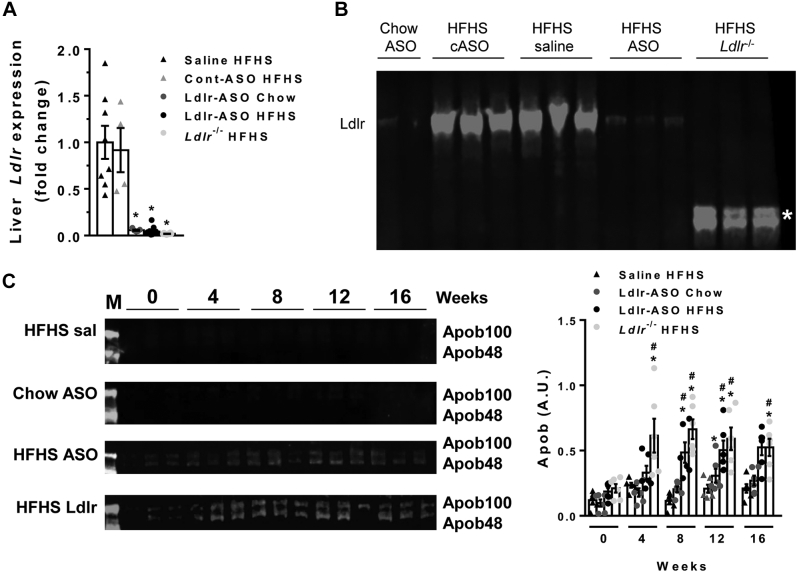
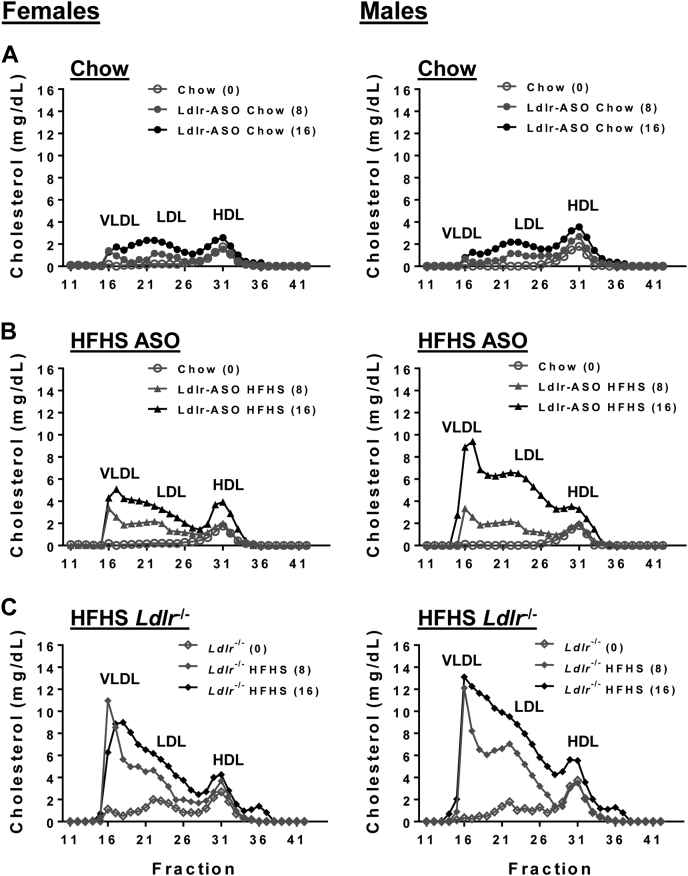
Antisense oligonucleotides (ASOs) against Ldl receptor (Ldlr-ASO) represent a promising strategy to promote hypercholesterolemic atherosclerosis in animal models without the need for complex breeding strategies. Here, we sought to characterize and contrast atherosclerosis in mice given Ldlr-ASO with those bearing genetic Ldlr deficiency. To promote atherosclerosis, male and female C57Bl6/J mice were either given weekly injections of Ldlr-ASO (5 mg/kg once per week) or genetically deficient in Ldlr (Ldlr-/-). Mice consumed either standard rodent chow or a diet high in saturated fat and sucrose with 0.15% added cholesterol for 16 weeks. While both models of Ldlr deficiency promoted hypercholesterolemia, Ldlr-/- mice exhibited nearly 2-fold higher cholesterol levels than Ldlr-ASO mice, reflected by increased VLDL and LDL levels. Consistent with this, the en face atherosclerotic lesion area was 3-fold and 3.6-fold greater in male and female mice with genetic Ldlr deficiency, respectively, as compared with the modest atherosclerosis observed following Ldlr-ASO treatment. Aortic sinus lesion sizes, fibrosis, smooth muscle actin, and necrotic core areas were also larger in Ldlr-/- mice, suggesting a more advanced phenotype. Despite a more modest effect on hypercholesterolemia, Ldlr-ASO induced greater hepatic inflammatory gene expression, macrophage accumulation, and histological lobular inflammation than was observed in Ldlr-/- mice. We conclude Ldlr-ASO is a promising tool for the generation of complex rodent models with which to study atherosclerosis but does not promote comparable levels of hypercholesterolemia or atherosclerosis as Ldlr-/- mice and increases hepatic inflammation. Thus, genetic Ldlr deficiency may be a superior model, depending on the proposed use.
Keywords: animal models; antisense oligonucleotides; fast-phase liquid chromatography; hepatic inflammation; hyperlipidemia; inflammation; liver; receptors/lipoprotein; serum amyloid A.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures




References
-
- Zhang S.H., Reddick R.L., Piedrahita J.A., Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. - PubMed
-
- Bennett C.F., Swayze E.E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. - PubMed
-
- Tromp T.R., Stroes E.S.G., Hovingh G.K. Gene-based therapy in lipid management: the winding road from promise to practice. Expert Opin. Investig. Drugs. 2020;29:483–493. - PubMed
-
- Kastelein J.J., Wedel M.K., Baker B.F., Su J., Bradley J.D., Yu R.Z., Chuang E., Graham M.J., Crooke R.M. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114:1729–1735. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
