

Histone acetylome-wide associations in immune cells from individuals with active Mycobacterium tuberculosis infection
- PMID: 35102304
- PMCID: PMC9439955
- DOI: 10.1038/s41564-021-01049-w
Histone acetylome-wide associations in immune cells from individuals with active Mycobacterium tuberculosis infection
Erratum in
-
Author Correction: Histone acetylome-wide associations in immune cells from individuals with active Mycobacterium tuberculosis infection.Nat Microbiol. 2022 Nov;7(11):1943. doi: 10.1038/s41564-022-01236-3. Nat Microbiol. 2022. PMID: 36056160 Free PMC article. No abstract available.
Abstract
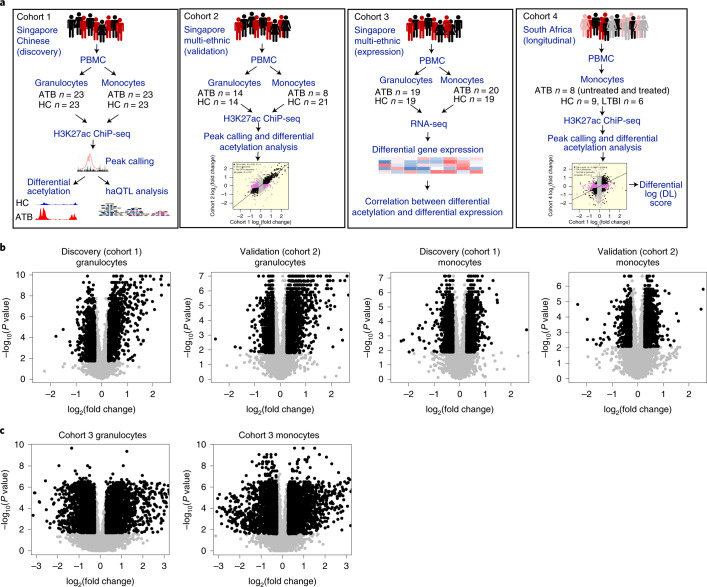
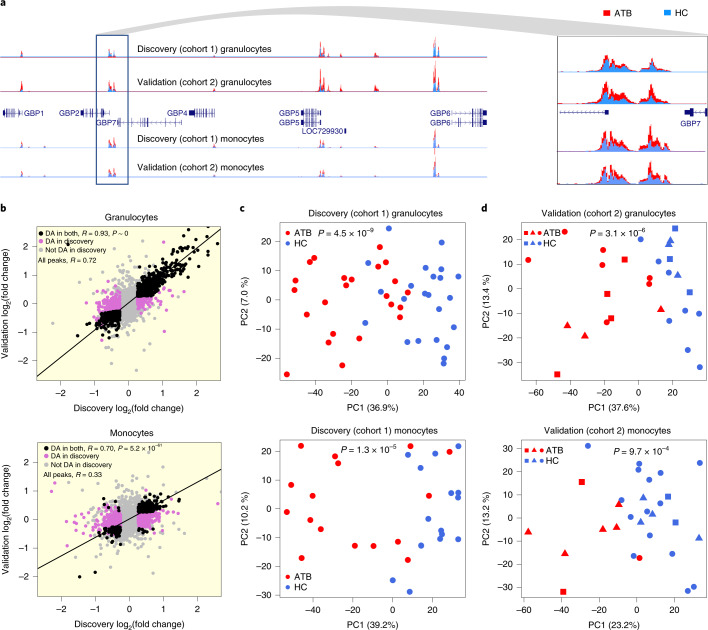
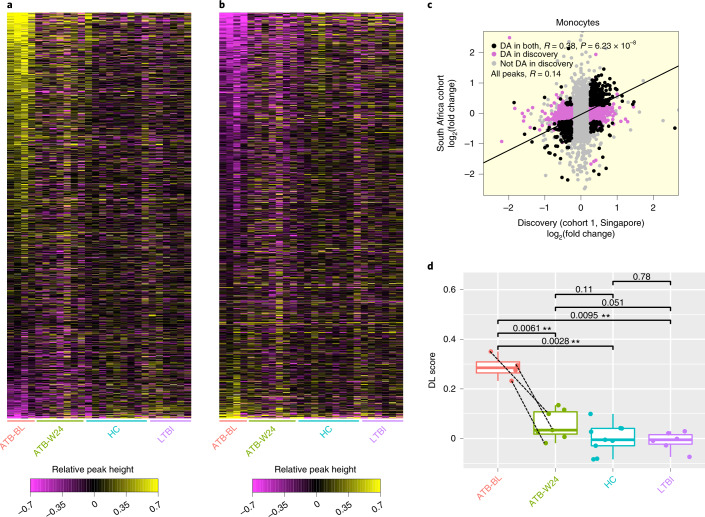
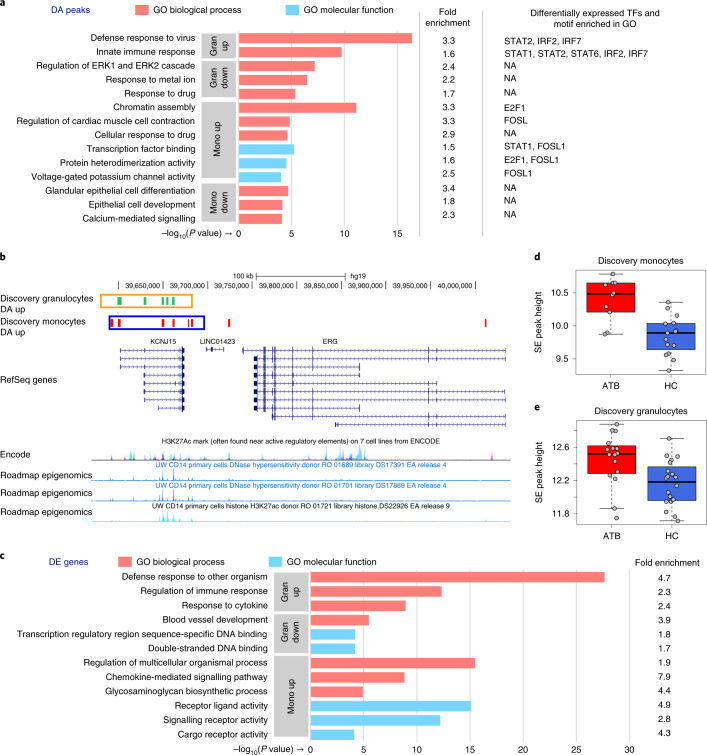
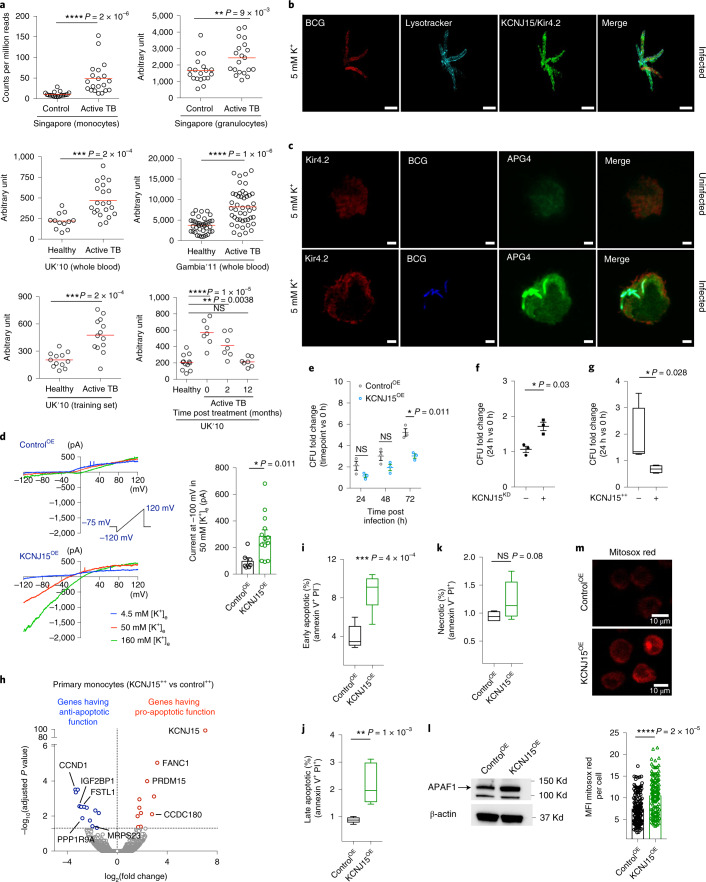
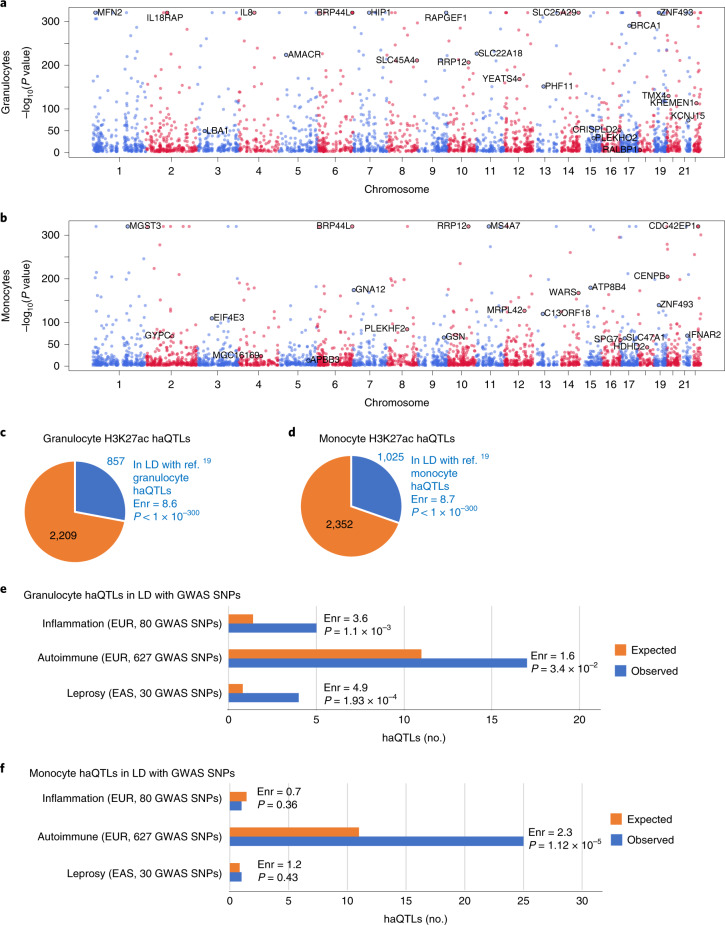
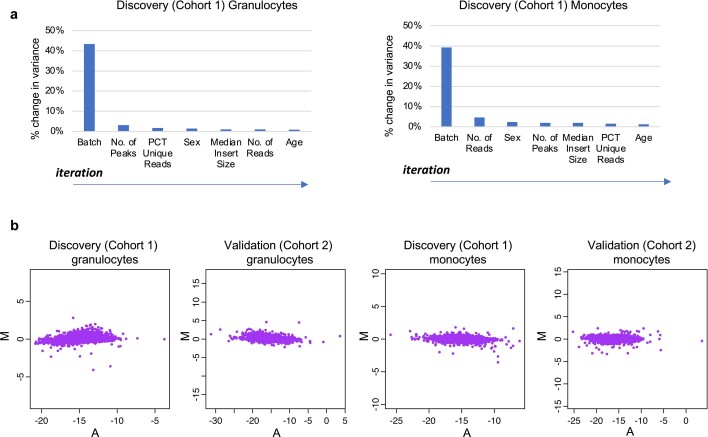
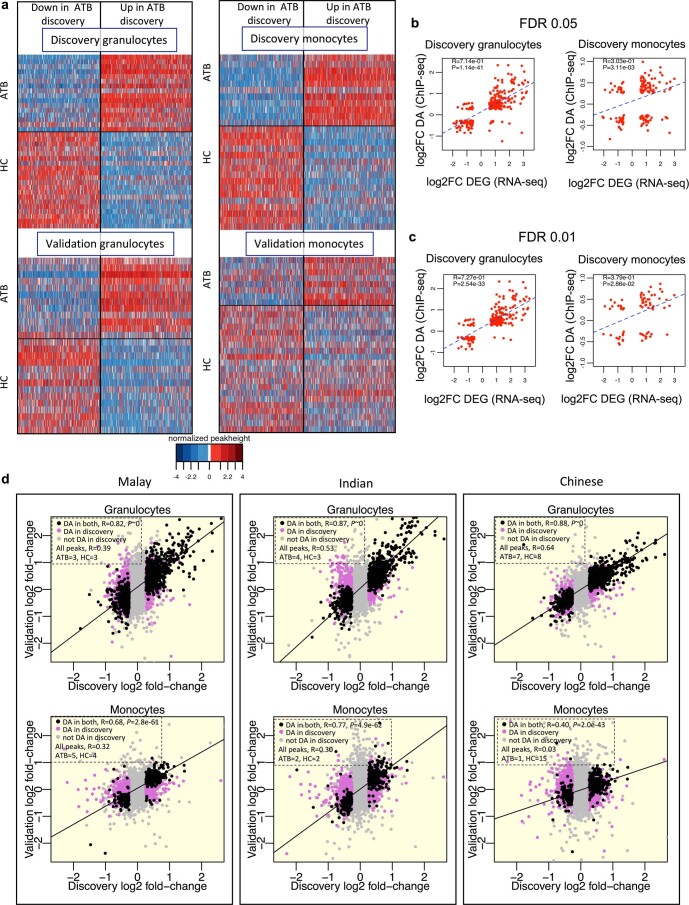
Host cell chromatin changes are thought to play an important role in the pathogenesis of infectious diseases. Here we describe a histone acetylome-wide association study (HAWAS) of an infectious disease, on the basis of genome-wide H3K27 acetylation profiling of peripheral blood granulocytes and monocytes from persons with active Mycobacterium tuberculosis (Mtb) infection and healthy controls. We detected >2,000 differentially acetylated loci in either cell type in a Singapore Chinese discovery cohort (n = 46), which were validated in a subsequent multi-ethnic Singapore cohort (n = 29), as well as a longitudinal cohort from South Africa (n = 26), thus demonstrating that HAWAS can be independently corroborated. Acetylation changes were correlated with differential gene expression. Differential acetylation was enriched near potassium channel genes, including KCNJ15, which modulates apoptosis and promotes Mtb clearance in vitro. We performed histone acetylation quantitative trait locus (haQTL) analysis on the dataset and identified 69 candidate causal variants for immune phenotypes among granulocyte haQTLs and 83 among monocyte haQTLs. Our study provides proof-of-principle for HAWAS to infer mechanisms of host response to pathogens.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
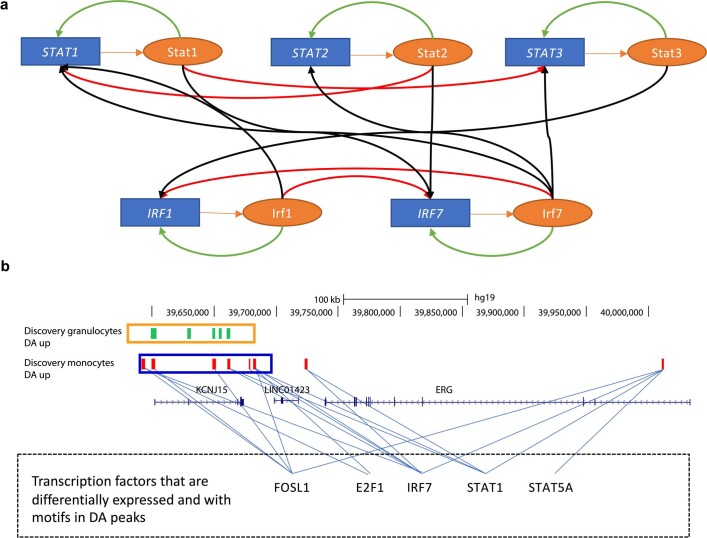
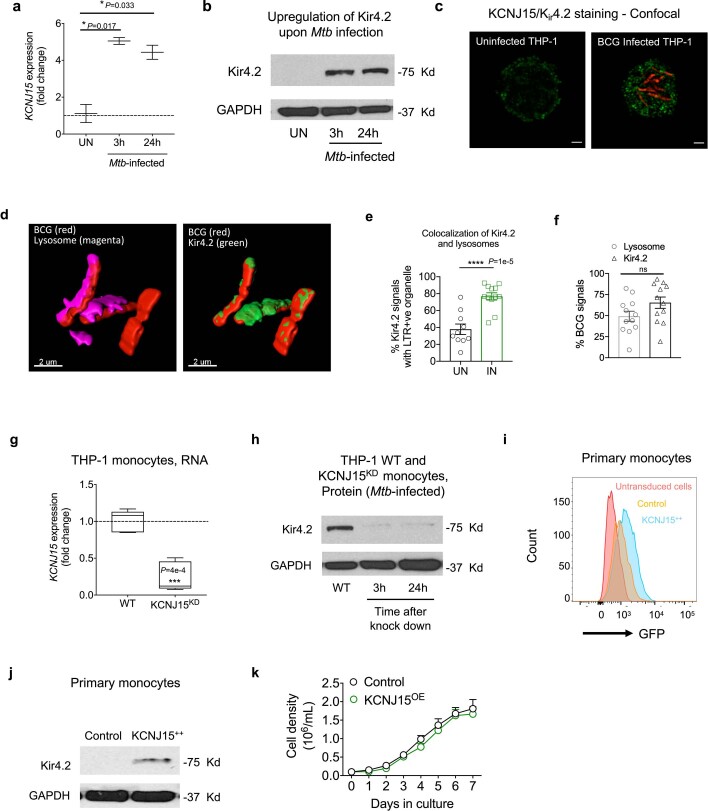
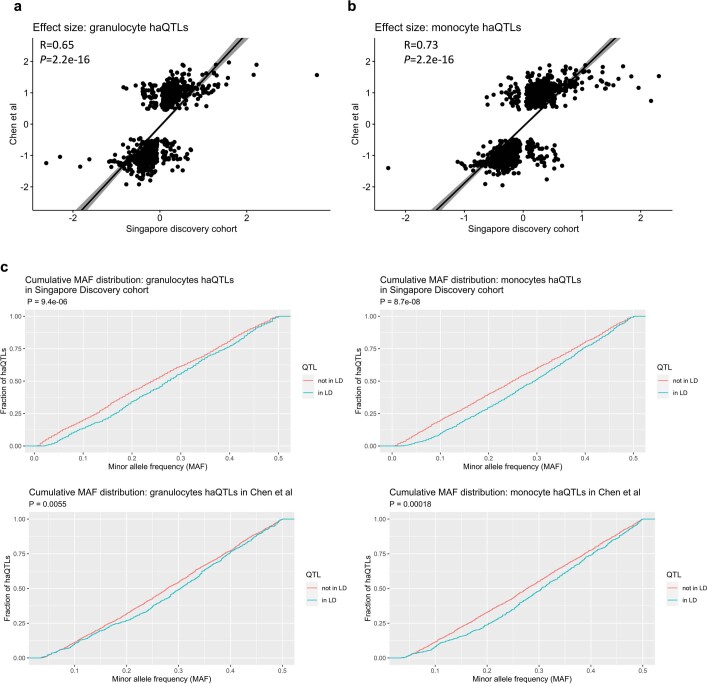
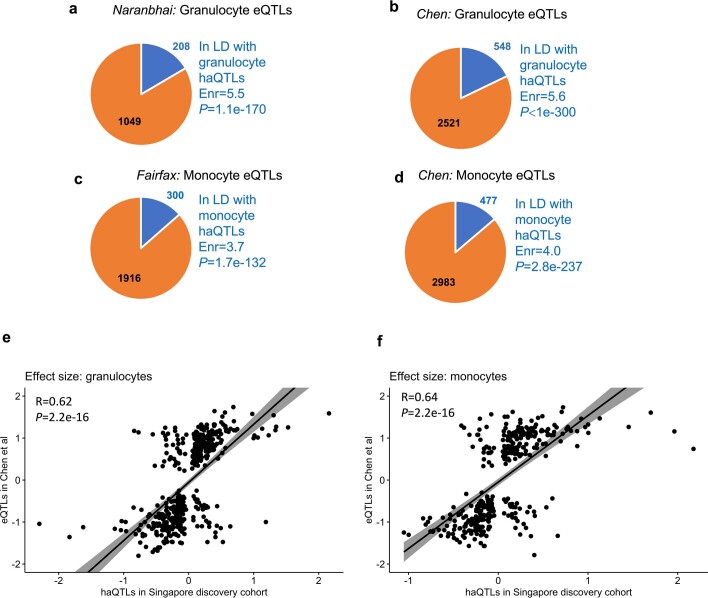
Comment in
-
Mycobacterium tuberculosis remodels host transcriptome.Nat Microbiol. 2022 Feb;7(2):189-190. doi: 10.1038/s41564-021-01056-x. Nat Microbiol. 2022. PMID: 35102305 No abstract available.
References
-
- WHO World Health Statistics 2016: Monitoring Health for the SDGshttp://www.who.int/gho/publications/world_health_statistics/2016/en/ (2016).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
