

Temporal modelling using single-cell transcriptomics
- PMID: 35102309
- PMCID: PMC10354343
- DOI: 10.1038/s41576-021-00444-7
Temporal modelling using single-cell transcriptomics
Abstract
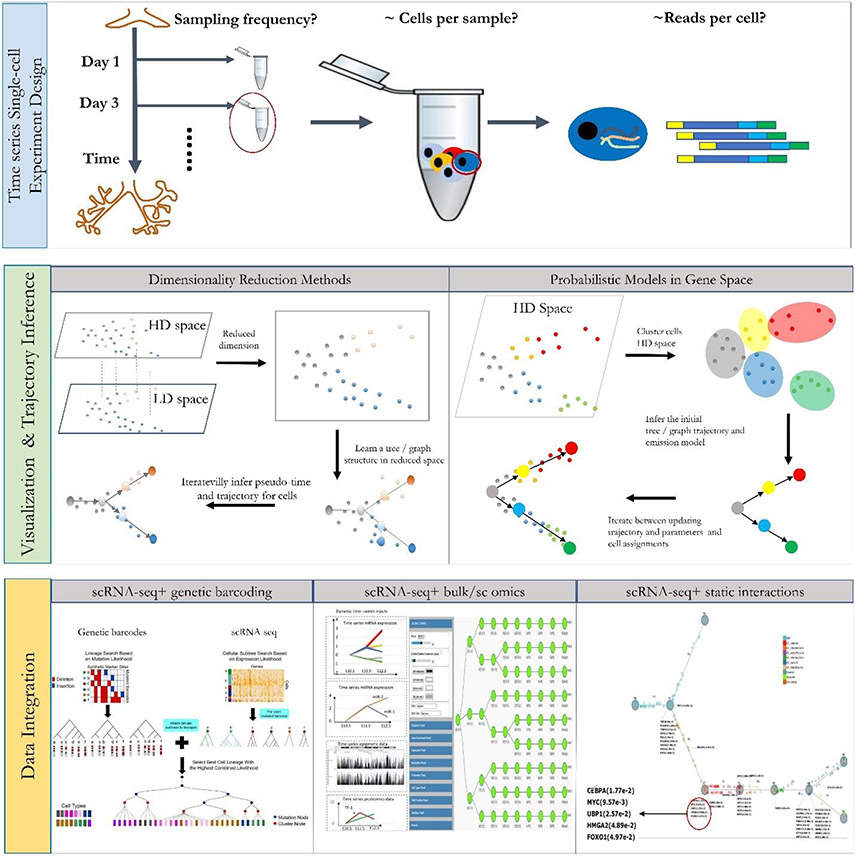
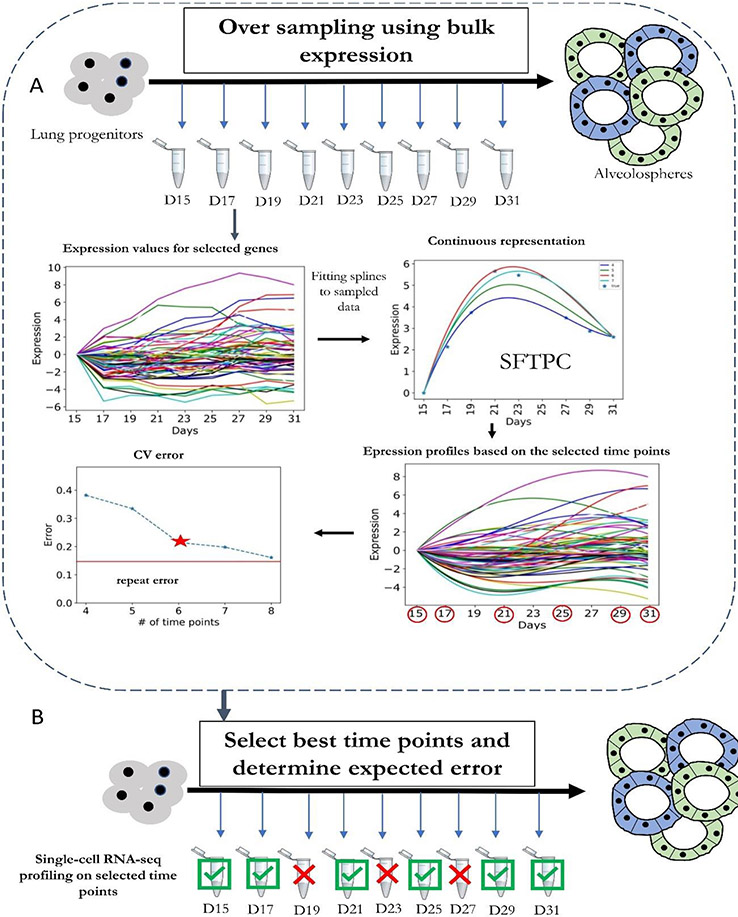
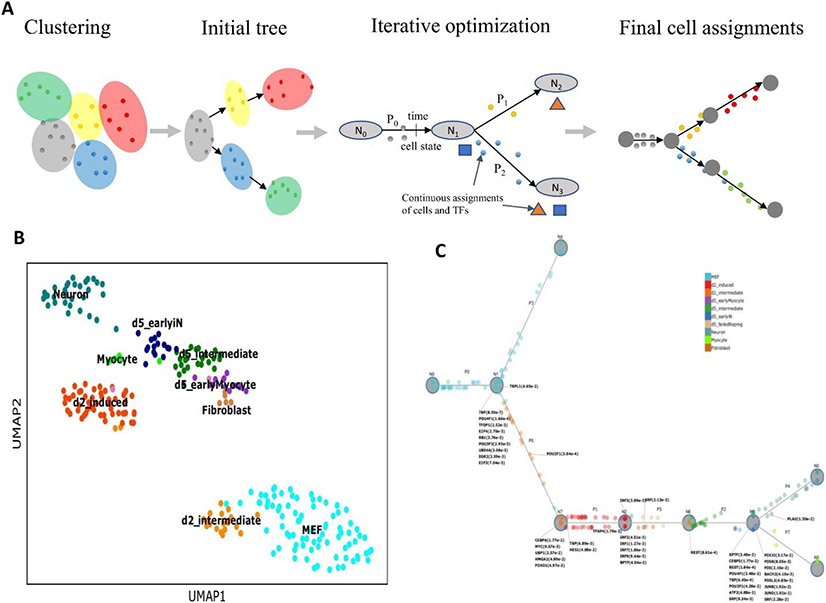
Methods for profiling genes at the single-cell level have revolutionized our ability to study several biological processes and systems including development, differentiation, response programmes and disease progression. In many of these studies, cells are profiled over time in order to infer dynamic changes in cell states and types, sets of expressed genes, active pathways and key regulators. However, time-series single-cell RNA sequencing (scRNA-seq) also raises several new analysis and modelling issues. These issues range from determining when and how deep to profile cells, linking cells within and between time points, learning continuous trajectories, and integrating bulk and single-cell data for reconstructing models of dynamic networks. In this Review, we discuss several approaches for the analysis and modelling of time-series scRNA-seq, highlighting their steps, key assumptions, and the types of data and biological questions they are most appropriate for.
© 2022. Springer Nature Limited.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
