

Context-dependent induction of autoimmunity by TNF signaling deficiency
- PMID: 35104241
- PMCID: PMC8983147
- DOI: 10.1172/jci.insight.149094
Context-dependent induction of autoimmunity by TNF signaling deficiency
Abstract
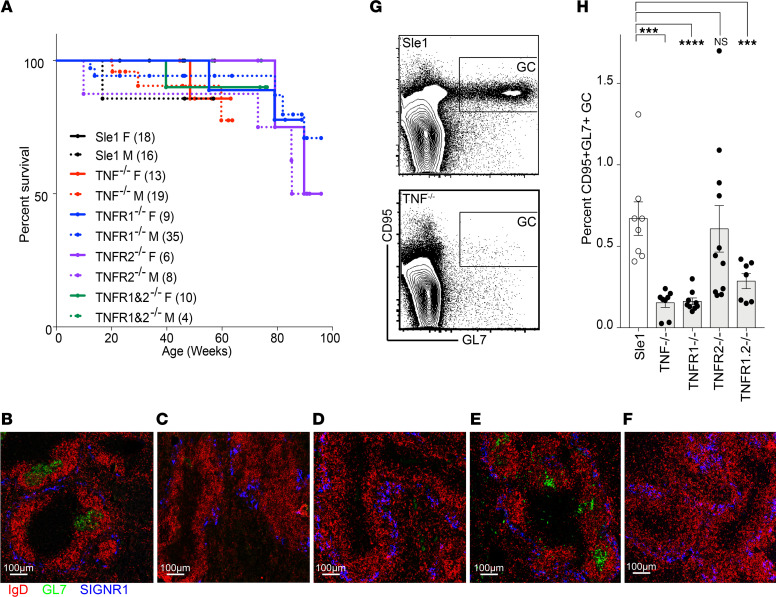
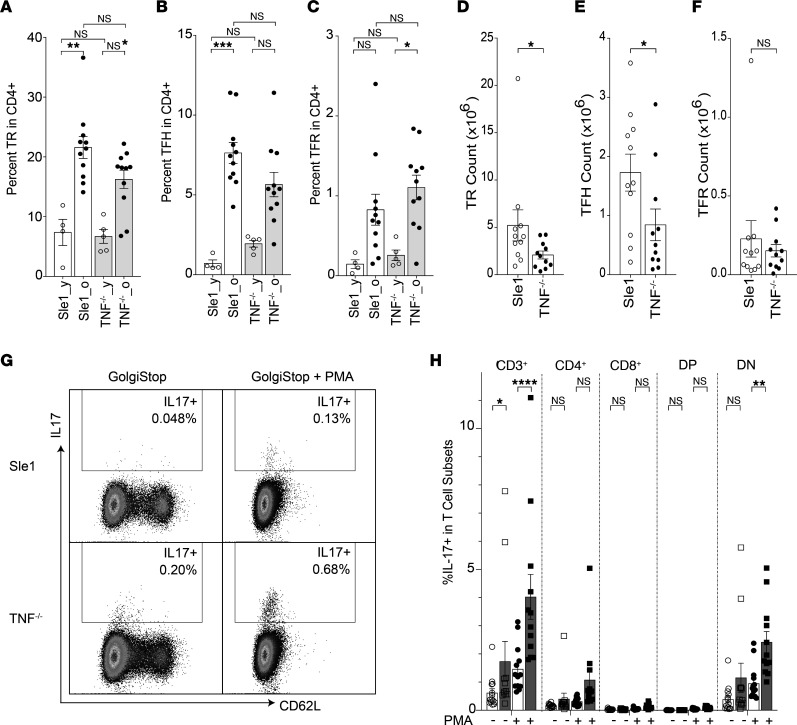
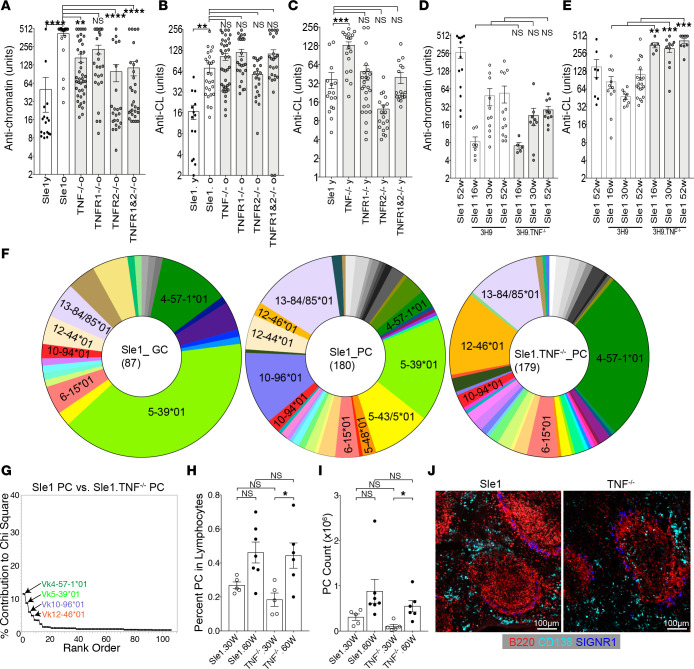
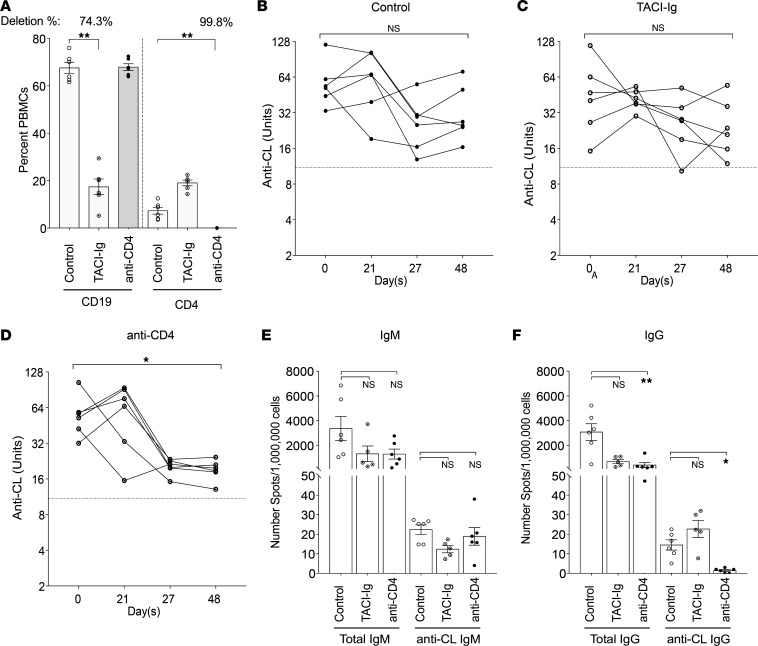
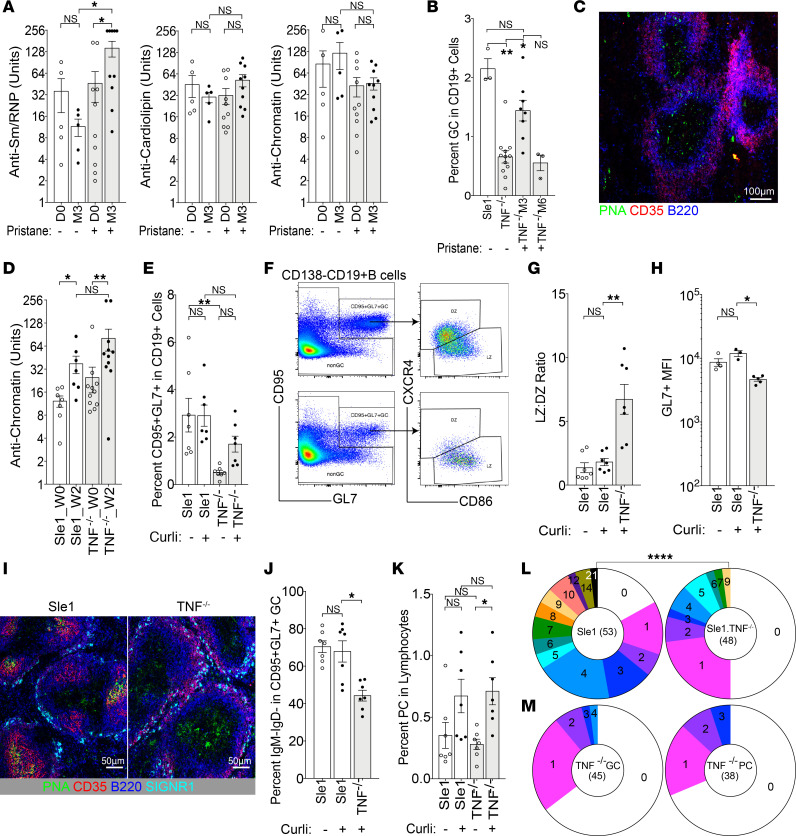
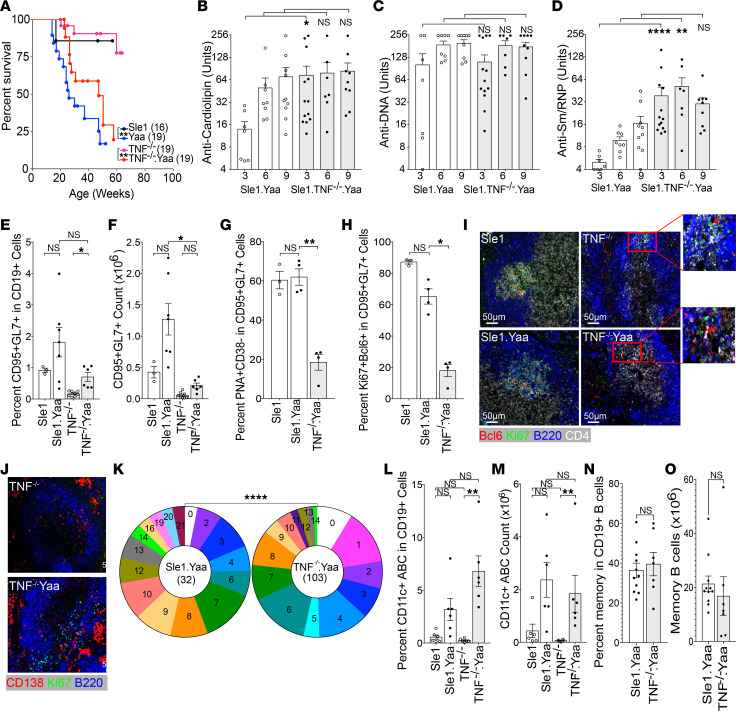
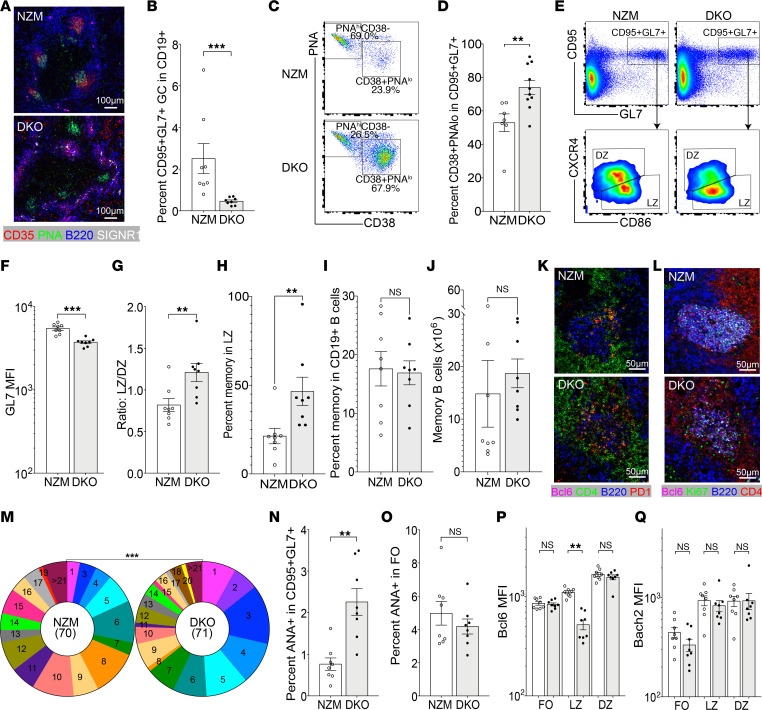
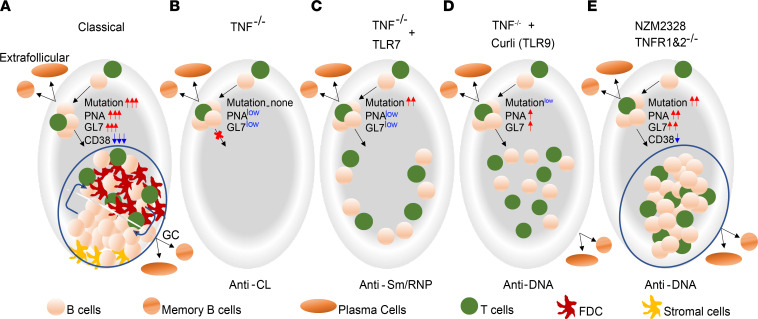
TNF inhibitors are widely used to treat inflammatory diseases; however, 30%-50% of treated patients develop new autoantibodies, and 0.5%-1% develop secondary autoimmune diseases, including lupus. TNF is required for formation of germinal centers (GCs), the site where high-affinity autoantibodies are often made. We found that TNF deficiency in Sle1 mice induced TH17 T cells and enhanced the production of germline encoded, T-dependent IgG anti-cardiolipin antibodies but did not induce GC formation or precipitate clinical disease. We then asked whether a second hit could restore GC formation or induce pathogenic autoimmunity in TNF-deficient mice. By using a range of immune stimuli, we found that somatically mutated autoantibodies and clinical disease can arise in the setting of TNF deficiency via extrafollicular pathways or via atypical GC-like pathways. This breach of tolerance may be due to defects in regulatory signals that modulate the negative selection of pathogenic autoreactive B cells.
Keywords: Autoimmune diseases; Immunology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
