

PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation
- PMID: 35105861
- PMCID: PMC8807747
- DOI: 10.1038/s41467-022-28198-8
PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation
Abstract
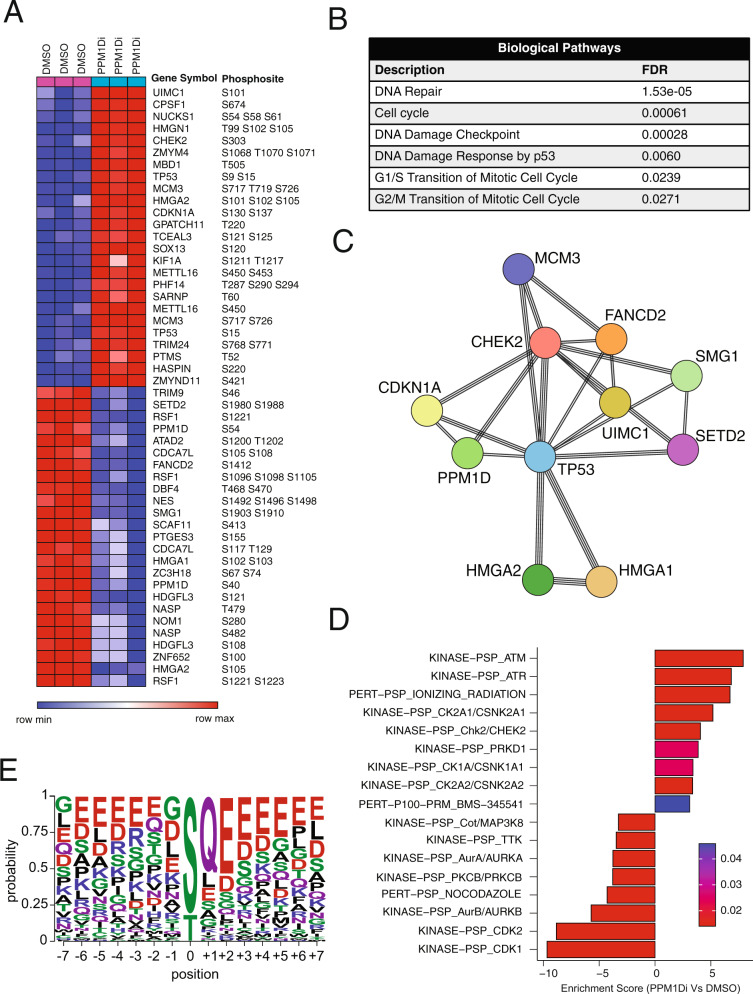
The role of PPM1D mutations in de novo gliomagenesis has not been systematically explored. Here we analyze whole genome sequences of 170 pediatric high-grade gliomas and find that truncating mutations in PPM1D that increase the stability of its phosphatase are clonal driver events in 11% of Diffuse Midline Gliomas (DMGs) and are enriched in primary pontine tumors. Through the development of DMG mouse models, we show that PPM1D mutations potentiate gliomagenesis and that PPM1D phosphatase activity is required for in vivo oncogenesis. Finally, we apply integrative phosphoproteomic and functional genomics assays and find that oncogenic effects of PPM1D truncation converge on regulators of cell cycle, DNA damage response, and p53 pathways, revealing therapeutic vulnerabilities including MDM2 inhibition.
© 2022. The Author(s).
Conflict of interest statement
R.B. and P.B. receive grant funding from the Novartis Institute of Biomedical Research for an unrelated project. P.B. has received grant funding from Deerfield and reports a consulting role for QED Therapeutics, R.B. reports consulting or advisory role for Novartis, Merck (I), Gilead Sciences (I), ViiV Healthcare (I); research funding from Novartis; patents, royalties, other intellectual property—Prognostic Marker for Endometrial Carcinoma (US patent application 13/911456, filed June 6, 2013), SF3B1 Suppression as a Therapy for Tumors Harboring SF3B1 Copy Loss (international application No. WO/2017/177191, PCT/US2017/026693, filed July 4, 2017), Compositions and Methods for Screening Pediatric Gliomas and Methods of Treatment Thereof (international application No. WO/2017/132574, PCT/US2017/015448, filed 1/27/2017). M.W.K. is now an employee of Bristol-Myers Squibb. F.P. is now an employee of Merck Research Laboratories. S.A.C. is a member of the scientific advisory boards of Kymera, PTM BioLabs and Seer and a scientific advisor to Pfizer and Biogen. D.E.R. receives research funding from Abbvie, Jannsen, Merck, and Vir through the Functional Genomics Consortium and serves on the board of directors of Addgene. B.L.E. has received research funding from Celgene and Deerfield. He has also received consulting fees from GRAIL, and he serves on the scientific advisory boards for and holds equity in Skyhawk Therapeutics and Exo Therapeutics. (I) denotes a competing interest involving a first degree relative of the author. All the remaining authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
