

Comprehensive metabolomics expands precision medicine for triple-negative breast cancer
- PMID: 35105939
- PMCID: PMC9061756
- DOI: 10.1038/s41422-022-00614-0
Comprehensive metabolomics expands precision medicine for triple-negative breast cancer
Abstract
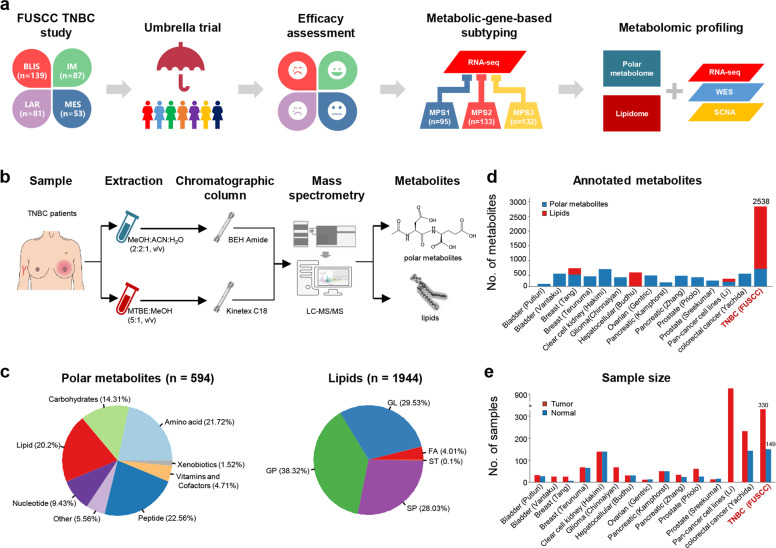
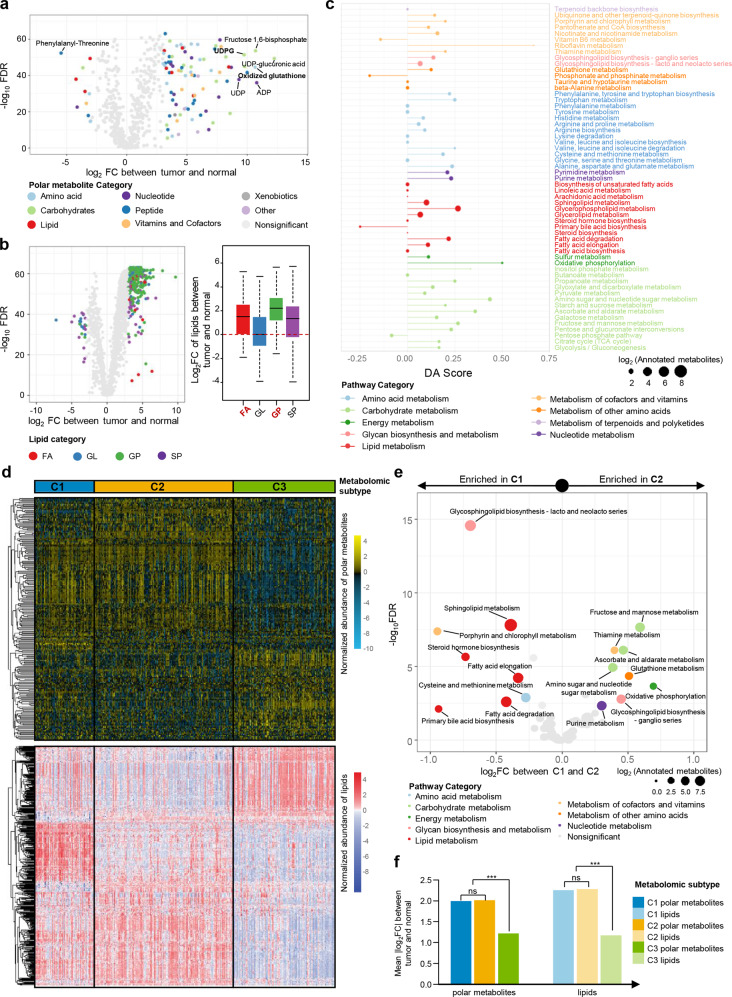
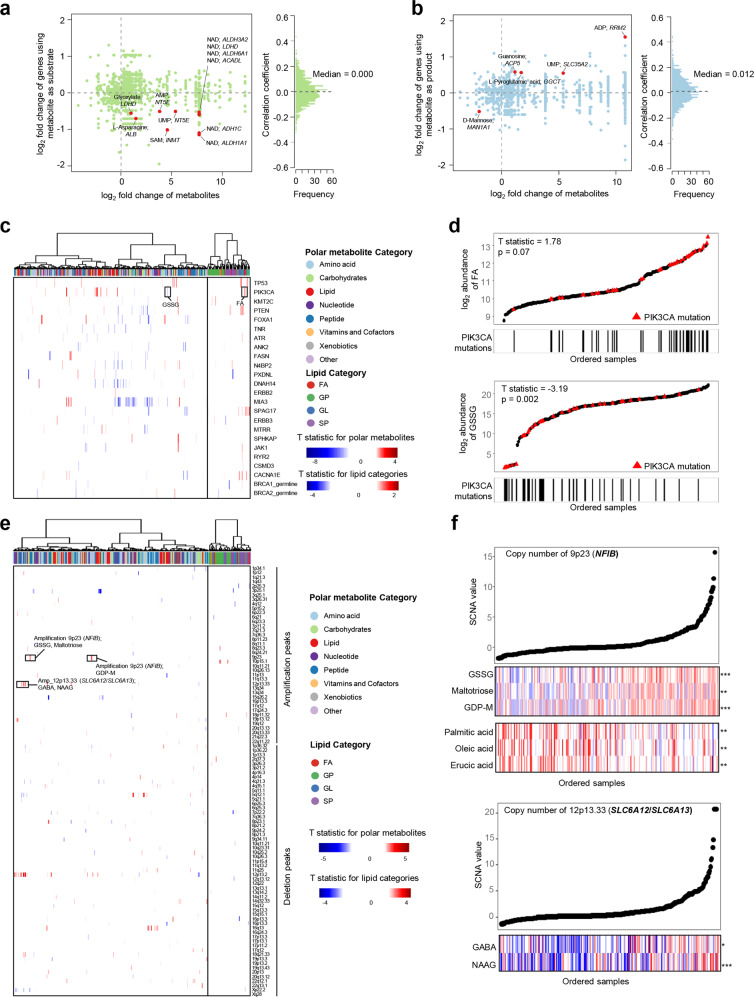
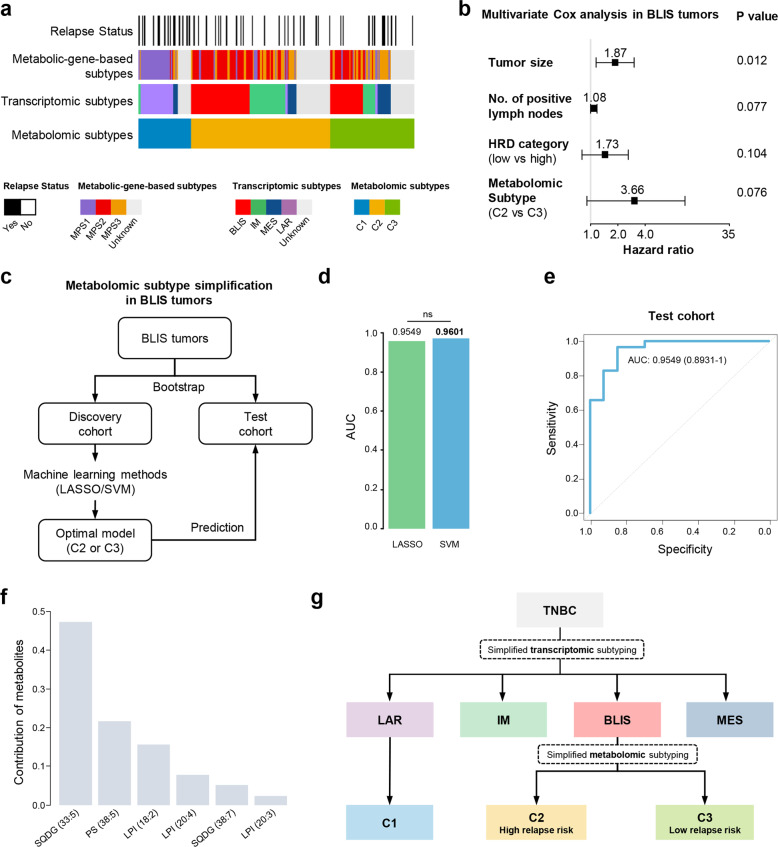
Metabolic reprogramming is a hallmark of cancer. However, systematic characterizations of metabolites in triple-negative breast cancer (TNBC) are still lacking. Our study profiled the polar metabolome and lipidome in 330 TNBC samples and 149 paired normal breast tissues to construct a large metabolomic atlas of TNBC. Combining with previously established transcriptomic and genomic data of the same cohort, we conducted a comprehensive analysis linking TNBC metabolome to genomics. Our study classified TNBCs into three distinct metabolomic subgroups: C1, characterized by the enrichment of ceramides and fatty acids; C2, featured with the upregulation of metabolites related to oxidation reaction and glycosyl transfer; and C3, having the lowest level of metabolic dysregulation. Based on this newly developed metabolomic dataset, we refined previous TNBC transcriptomic subtypes and identified some crucial subtype-specific metabolites as potential therapeutic targets. The transcriptomic luminal androgen receptor (LAR) subtype overlapped with metabolomic C1 subtype. Experiments on patient-derived organoid and xenograft models indicate that targeting sphingosine-1-phosphate (S1P), an intermediate of the ceramide pathway, is a promising therapy for LAR tumors. Moreover, the transcriptomic basal-like immune-suppressed (BLIS) subtype contained two prognostic metabolomic subgroups (C2 and C3), which could be distinguished through machine-learning methods. We show that N-acetyl-aspartyl-glutamate is a crucial tumor-promoting metabolite and potential therapeutic target for high-risk BLIS tumors. Together, our study reveals the clinical significance of TNBC metabolomics, which can not only optimize the transcriptomic subtyping system, but also suggest novel therapeutic targets. This metabolomic dataset can serve as a useful public resource to promote precision treatment of TNBC.
Trial registration: ClinicalTrials.gov NCT03805399.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


Comment in
-
Banking on metabolomics for novel therapies in TNBC.Cell Res. 2022 May;32(5):423-424. doi: 10.1038/s41422-022-00637-7. Cell Res. 2022. PMID: 35228657 Free PMC article. No abstract available.
References
-
- Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389:2430–2442. - PubMed
-
- Jiang YZ, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. 2019;35:428–440 e5. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
