

Identification of neoantigens for individualized therapeutic cancer vaccines
- PMID: 35105974
- PMCID: PMC7612664
- DOI: 10.1038/s41573-021-00387-y
Identification of neoantigens for individualized therapeutic cancer vaccines
Erratum in
-
Author Correction: Identification of neoantigens for individualized therapeutic cancer vaccines.Nat Rev Drug Discov. 2024 Feb;23(2):156. doi: 10.1038/s41573-023-00873-5. Nat Rev Drug Discov. 2024. PMID: 38086950 No abstract available.
Abstract
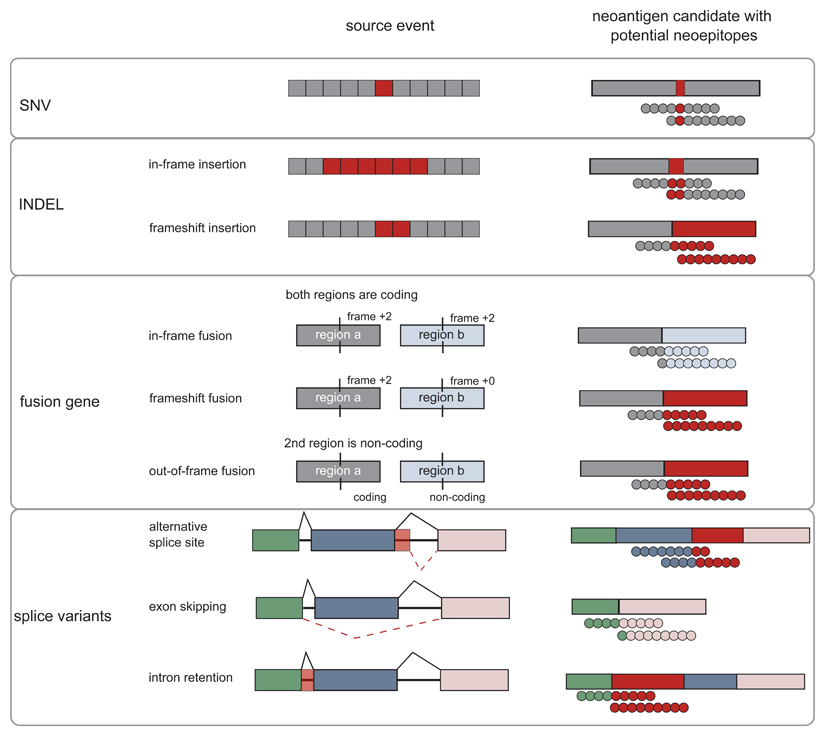
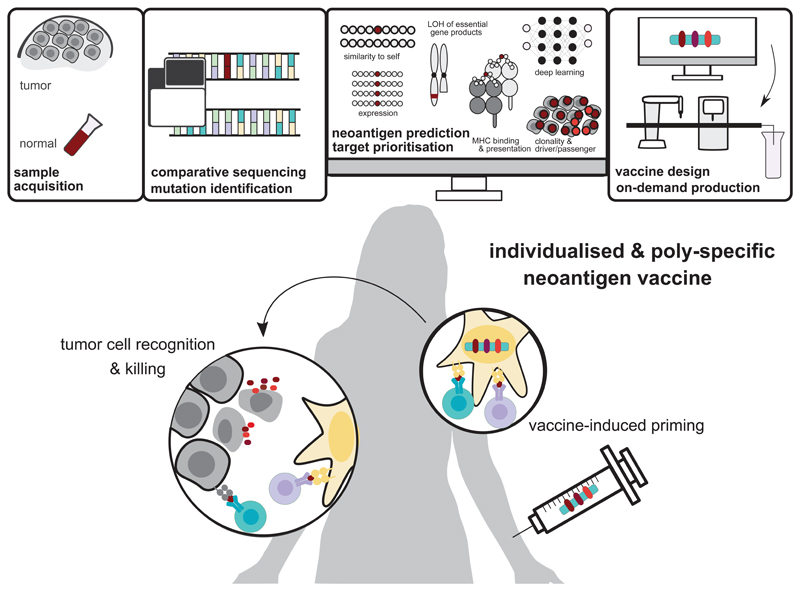
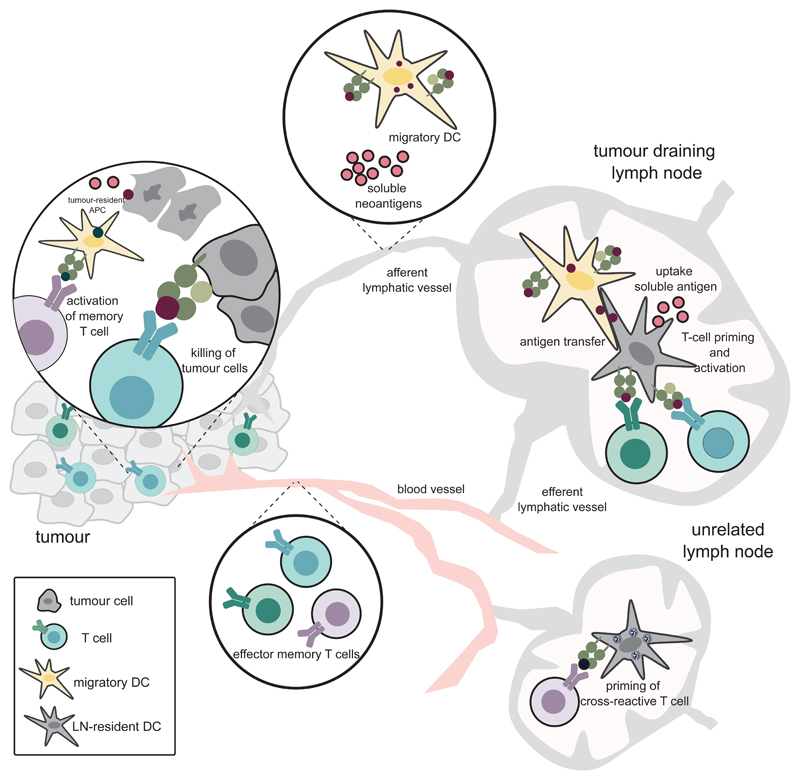
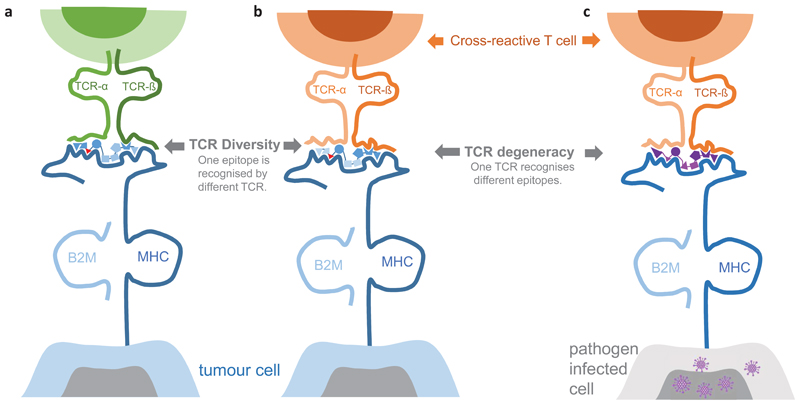
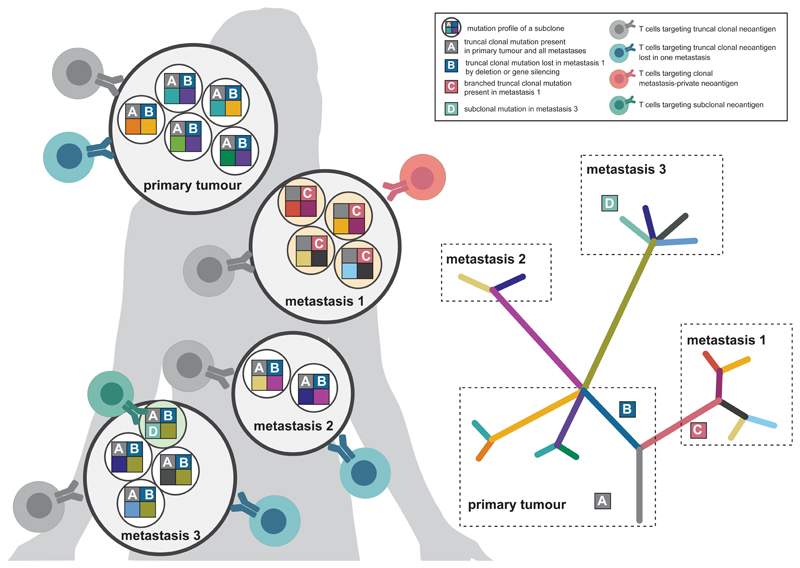
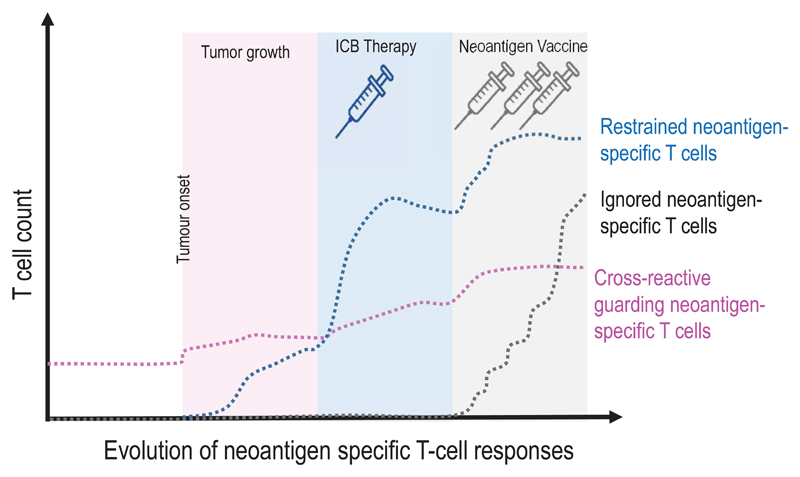
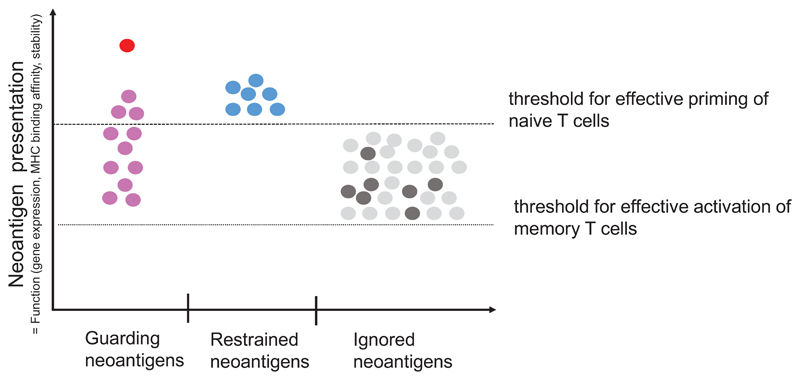
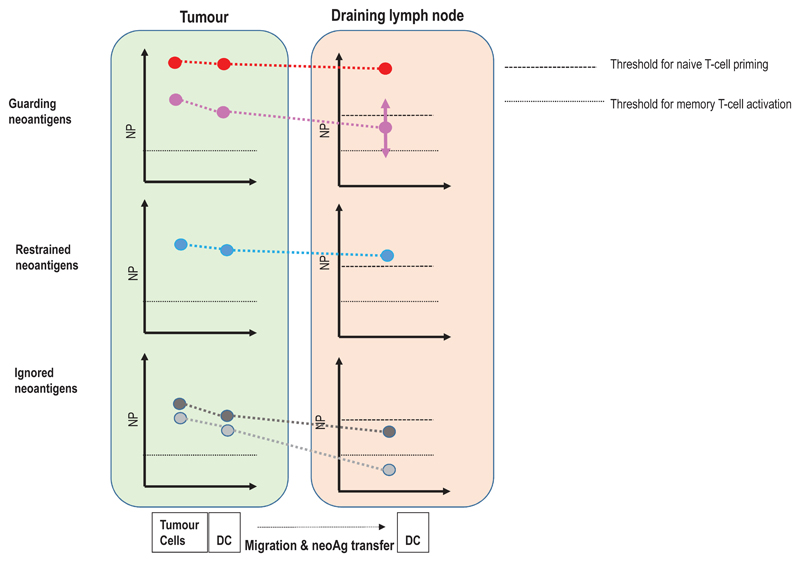
Somatic mutations in cancer cells can generate tumour-specific neoepitopes, which are recognized by autologous T cells in the host. As neoepitopes are not subject to central immune tolerance and are not expressed in healthy tissues, they are attractive targets for therapeutic cancer vaccines. Because the vast majority of cancer mutations are unique to the individual patient, harnessing the full potential of this rich source of targets requires individualized treatment approaches. Many computational algorithms and machine-learning tools have been developed to identify mutations in sequence data, to prioritize those that are more likely to be recognized by T cells and to design tailored vaccines for every patient. In this Review, we fill the gaps between the understanding of basic mechanisms of T cell recognition of neoantigens and the computational approaches for discovery of somatic mutations and neoantigen prediction for cancer immunotherapy. We present a new classification of neoantigens, distinguishing between guarding, restrained and ignored neoantigens, based on how they confer proficient antitumour immunity in a given clinical context. Such context-based differentiation will contribute to a framework that connects neoantigen biology to the clinical setting and medical peculiarities of cancer, and will enable future neoantigen-based therapies to provide greater clinical benefit.
© 2022. Springer Nature Limited.
Conflict of interest statement
F.L. has nothing to disclose. B.S., M.L., Ö.T. and U.S. are inventors on patents related to some of the technologies described in this article. Ö.T. is shareholder and CMO at BioNTech SE. U.S. is co-founder, shareholder and CEO at BioNTech SE.
Figures
References
-
- Chiari R, et al. Two antigens recognized by autologous cytolytic T lymphocytes on a melanoma result from a single point mutation in an essential housekeeping gene. Cancer Research. 1999;59:5785–5792. - PubMed
Highlighted References
-
- Alexandrov Ludmil B, Nik-Zainal Serena, Wedge David C, Aparicio Samuel AJR, Behjati Sam, Biankin Andrew V, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [This article illustrates the prevalence of somatic point mutations across human cancers.] - DOI - PMC - PubMed
-
- Alspach Elise, Lussier Danielle M, Miceli Alexander P, Kizhvatov Ilya, DuPage Michel, Luoma Adrienne M, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574(7780):696–701. doi: 10.1038/s41586-019-1671-8. [This article underlines the relevance of MHC class II neoantigens for an efficient anti-tumour response.] - DOI - PMC - PubMed
-
- Balachandran Vinod P, Łuksza Marta, Zhao Julia N, Makarov Vladimir, Moral John Alec, Remark Romain, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512. doi: 10.1038/nature24462. [This article highlights that neoantigen quality correlates with clinical outcome.] - DOI - PMC - PubMed
-
- Rosenthal Rachel, Cadieux Elizabeth Larose, Salgado Roberto, Bakir Maise Al, Moore David A, Hiley Crispin T, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567(7749):479–485. doi: 10.1038/s41586-019-1032-7. [ This article integrates genomic features of tumours with immune infiltrates and analyses neoantigen-dependent immune escape. ] - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
