

Evolution of Viral Pathogens Follows a Linear Order
- PMID: 35107326
- PMCID: PMC8809352
- DOI: 10.1128/spectrum.01655-21
Evolution of Viral Pathogens Follows a Linear Order
Abstract
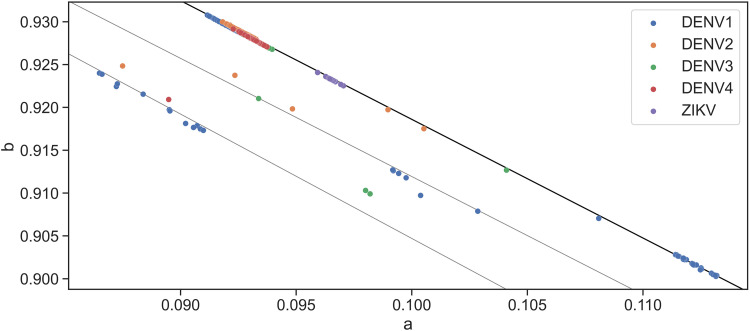
Although lessons have been learned from previous severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) outbreaks, the rapid evolution of the viruses means that future outbreaks of a much larger scale are possible, as shown by the current coronavirus disease 2019 (COVID-19) outbreak. Therefore, it is necessary to better understand the evolution of coronaviruses as well as viruses in general. This study reports a comparative analysis of the amino acid usage within several key viral families and genera that are prone to triggering outbreaks, including coronavirus (severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2], SARS-CoV, MERS-CoV, human coronavirus-HKU1 [HCoV-HKU1], HCoV-OC43, HCoV-NL63, and HCoV-229E), influenza A (H1N1 and H3N2), flavivirus (dengue virus serotypes 1 to 4 and Zika) and ebolavirus (Zaire, Sudan, and Bundibugyo ebolavirus). Our analysis reveals that the distribution of amino acid usage in the viral genome is constrained to follow a linear order, and the distribution remains closely related to the viral species within the family or genus. This constraint can be adapted to predict viral mutations and future variants of concern. By studying previous SARS and MERS outbreaks, we have adapted this naturally occurring pattern to determine that although pangolin plays a role in the outbreak of COVID-19, it may not be the sole agent as an intermediate animal. In addition to this study, our findings contribute to the understanding of viral mutations for subsequent development of vaccines and toward developing a model to determine the source of the outbreak. IMPORTANCE This study reports a comparative analysis of amino acid usage within several key viral genera that are prone to triggering outbreaks. Interestingly, there is evidence that the amino acid usage within the viral genomes is not random but in a linear order.
Keywords: SARS-CoV-2; infectious disease; linear order; microbiology; outbreak; viral pathogen.
Conflict of interest statement
The authors declare no conflict of interest.
Figures













References
-
- Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, Liu L, Shan H, Lei C, Hui DSC, Du B, Li L, Zeng G, Yuen K-Y, Chen R, Tang C, Wang T, Chen P, Xiang J, Li S, Wang J-L, Liang Z, Peng Y, Wei L, Liu Y, Hu Y-H, Peng P, Wang J-M, Liu J, Chen Z, Li G, Zheng Z, Qiu S, Luo J, Ye C, Zhu S, Zhong N, China Medical Treatment Expert Group for Covid-19 . 2020. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720. doi: 10.1056/NEJMoa2002032. - DOI - PMC - PubMed
-
- Zhu N, Zhang DY, Wang WL, Li XW, Yang B, Song JD, Zhao X, Huang BY, Shi WF, Lu RJ, Niu PH, Zhan FX, Ma XJ, Wang DY, Xu WB, Wu GZ, Gao GF, Tan WJ, China Novel Coronavirus Investigating and Research Team . 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. doi: 10.1056/NEJMoa2001017. - DOI - PMC - PubMed
-
- Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, Hu Y, Tao Z-W, Tian J-H, Pei Y-Y, Yuan M-L, Zhang Y-L, Dai F-H, Liu Y, Wang Q-M, Zheng J-J, Xu L, Holmes EC, Zhang Y-Z. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. doi: 10.1038/s41586-020-2008-3. - DOI - PMC - PubMed
-
- Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G-F, Shi Z-L. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi: 10.1038/s41586-020-2012-7. - DOI - PMC - PubMed
-
- Lu RJ, Zhao X, Li J, Niu PH, Yang B, Wu HL, Wang WL, Song H, Huang BY, Zhu N, Bi YH, Ma XJ, Zhan FX, Wang L, Hu T, Zhou H, Hu ZH, Zhou WM, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan JY, Xie ZH, Ma JM, Liu WJ, Wang DY, Xu WB, Holmes EC, Gao GF, Wu GZ, Chen WJ, Shi WF, Tan WJ. 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. doi: 10.1016/S0140-6736(20)30251-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
