

The Statistics of k-mers from a Sequence Undergoing a Simple Mutation Process Without Spurious Matches
- PMID: 35108101
- PMCID: PMC11978275
- DOI: 10.1089/cmb.2021.0431
The Statistics of k-mers from a Sequence Undergoing a Simple Mutation Process Without Spurious Matches
Abstract
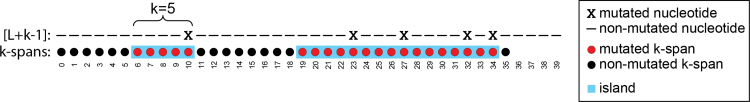
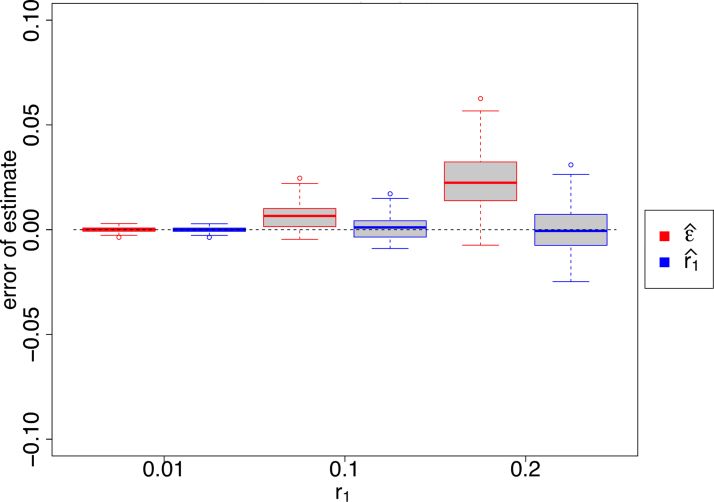
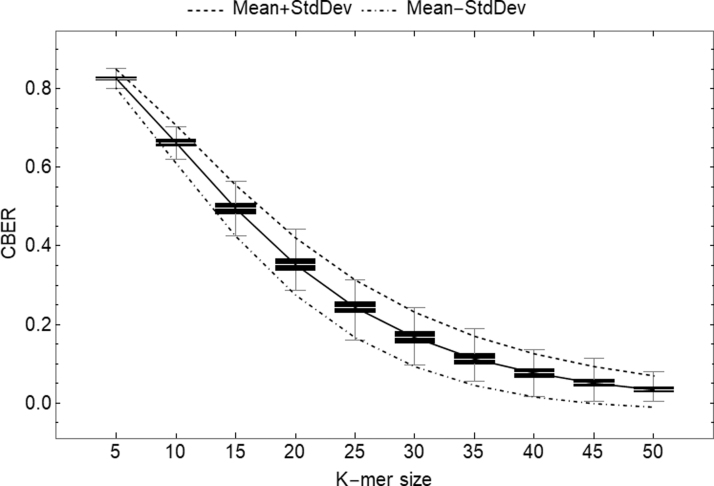
k-mer-based methods are widely used in bioinformatics, but there are many gaps in our understanding of their statistical properties. Here, we consider the simple model where a sequence S (e.g., a genome or a read) undergoes a simple mutation process through which each nucleotide is mutated independently with some probability r, under the assumption that there are no spurious k-mer matches. How does this process affect the k-mers of S? We derive the expectation and variance of the number of mutated k-mers and of the number of islands (a maximal interval of mutated k-mers) and oceans (a maximal interval of nonmutated k-mers). We then derive hypothesis tests and confidence intervals (CIs) for r given an observed number of mutated k-mers, or, alternatively, given the Jaccard similarity (with or without MinHash). We demonstrate the usefulness of our results using a few select applications: obtaining a CI to supplement the Mash distance point estimate, filtering out reads during alignment by Minimap2, and rating long-read alignments to a de Bruijn graph by Jabba.
Keywords: Jaccard similarity; MinHash; confidence intervals; k-mers; mutation process; sketching.
Figures
References
-
- Broder, A.Z. 1997. On the resemblance and containment of documents, pp. 21–29. In: Proceedings. Compression and Complexity of SEQUENCES 1997 (Cat. No. 97TB100171). IEEE, Salerno, Italy.
-
- Brown, L.D., Cai, T.T., and DasGupta, A.. 2001. Interval estimation for a binomial proportion. Stat. Sci. 16, 101–117.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
