

Global diversity and balancing selection of 23 leading Plasmodium falciparum candidate vaccine antigens
- PMID: 35108259
- PMCID: PMC8843232
- DOI: 10.1371/journal.pcbi.1009801
Global diversity and balancing selection of 23 leading Plasmodium falciparum candidate vaccine antigens
Abstract
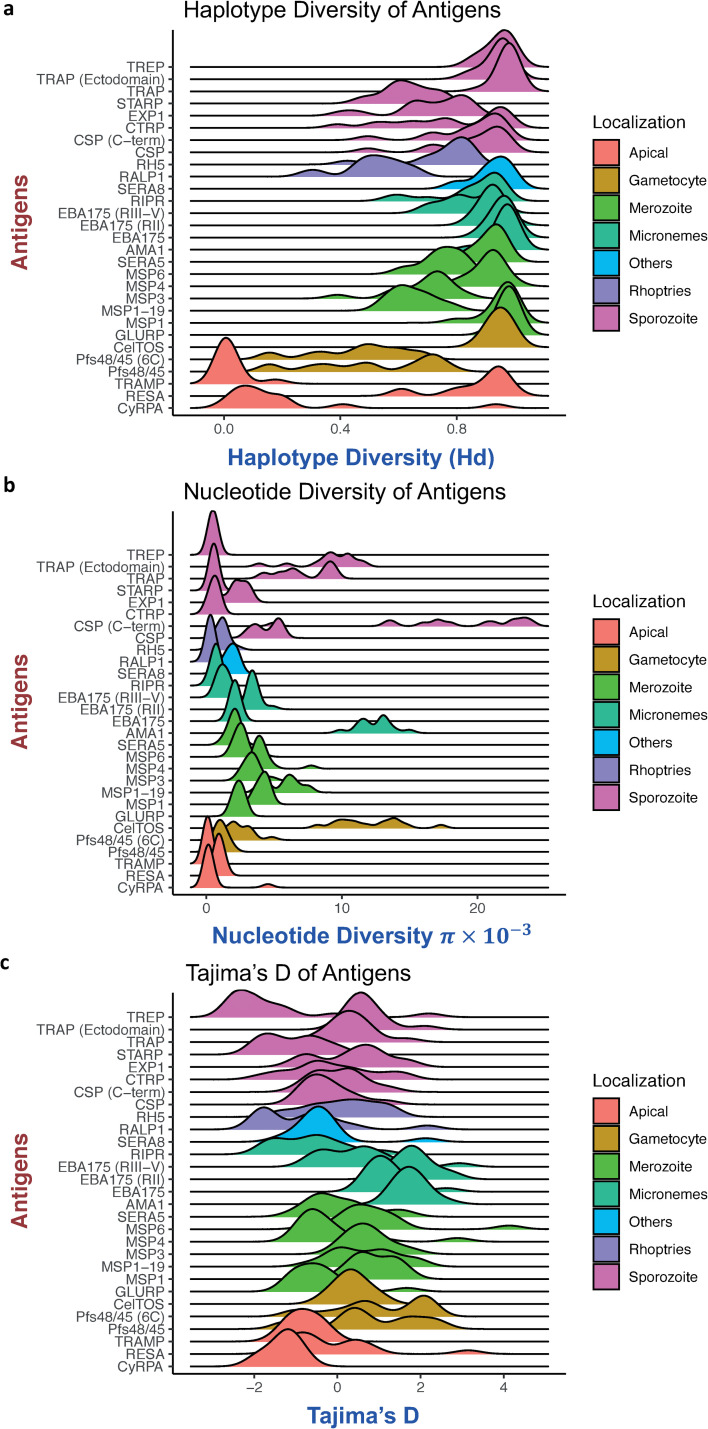
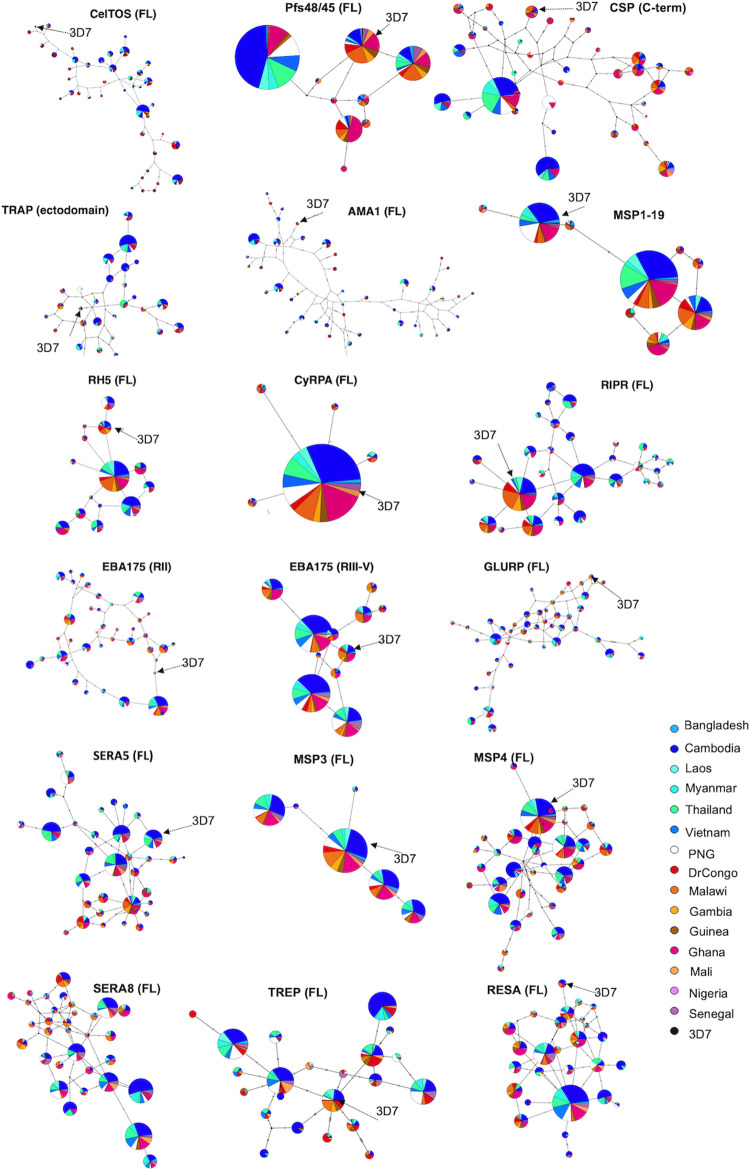
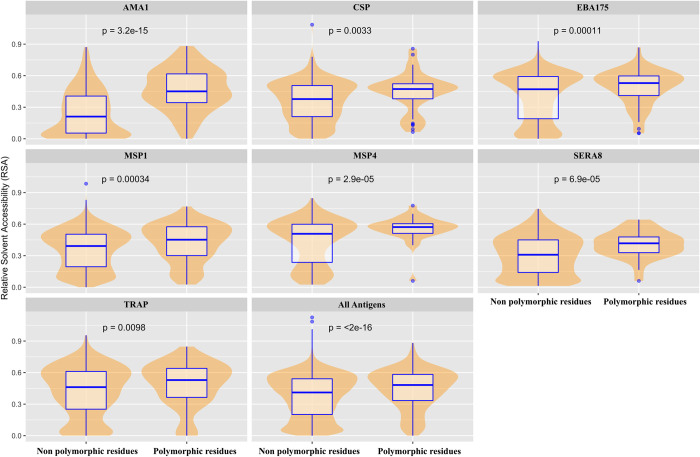
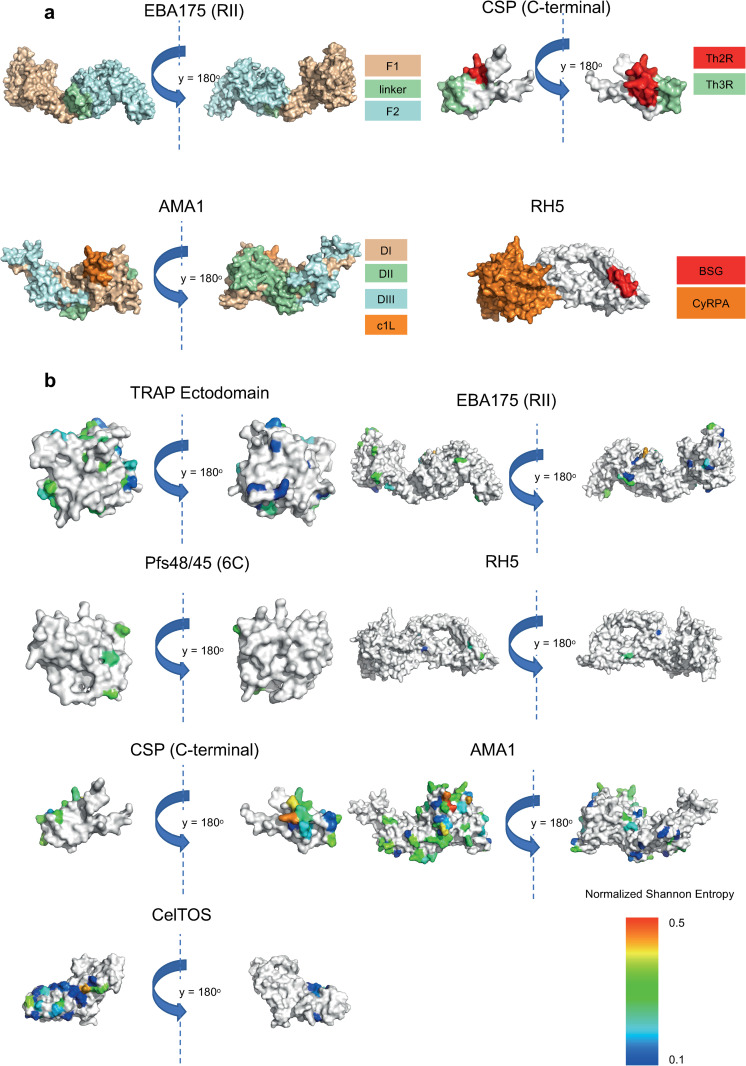
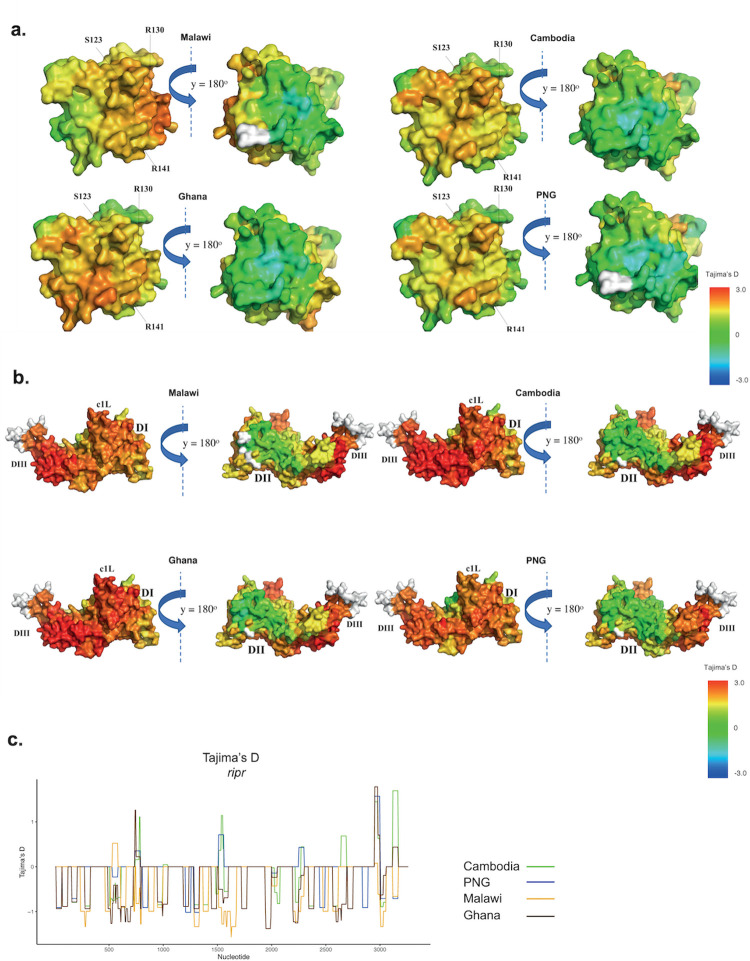
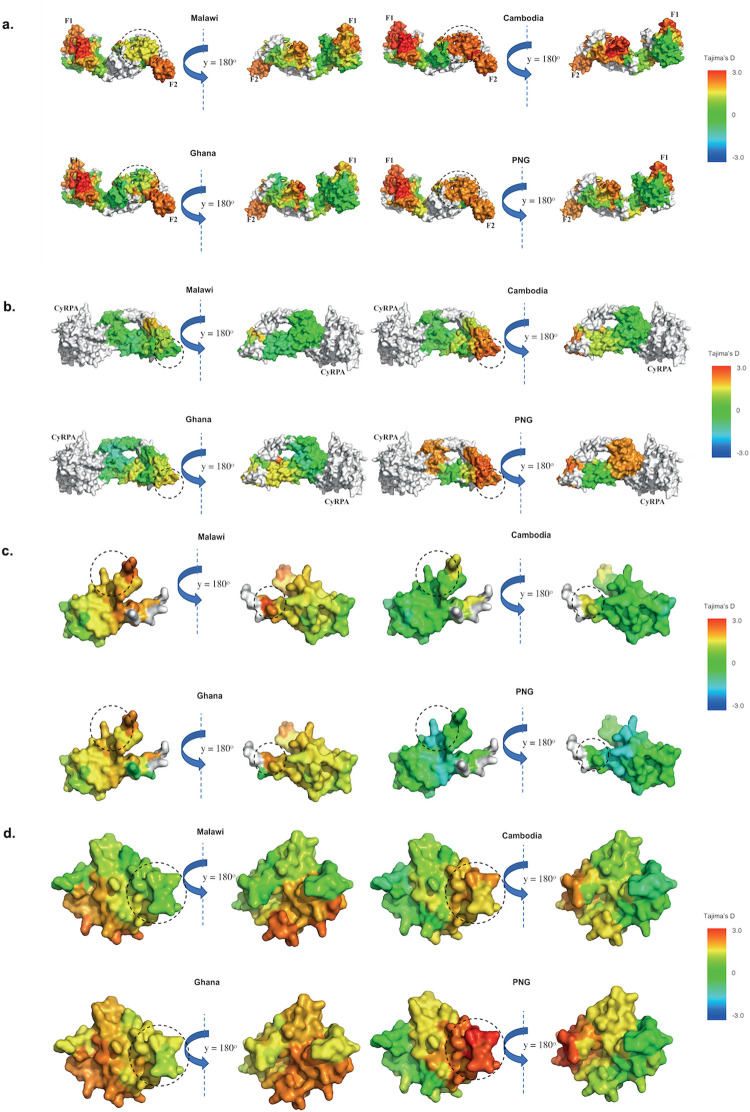
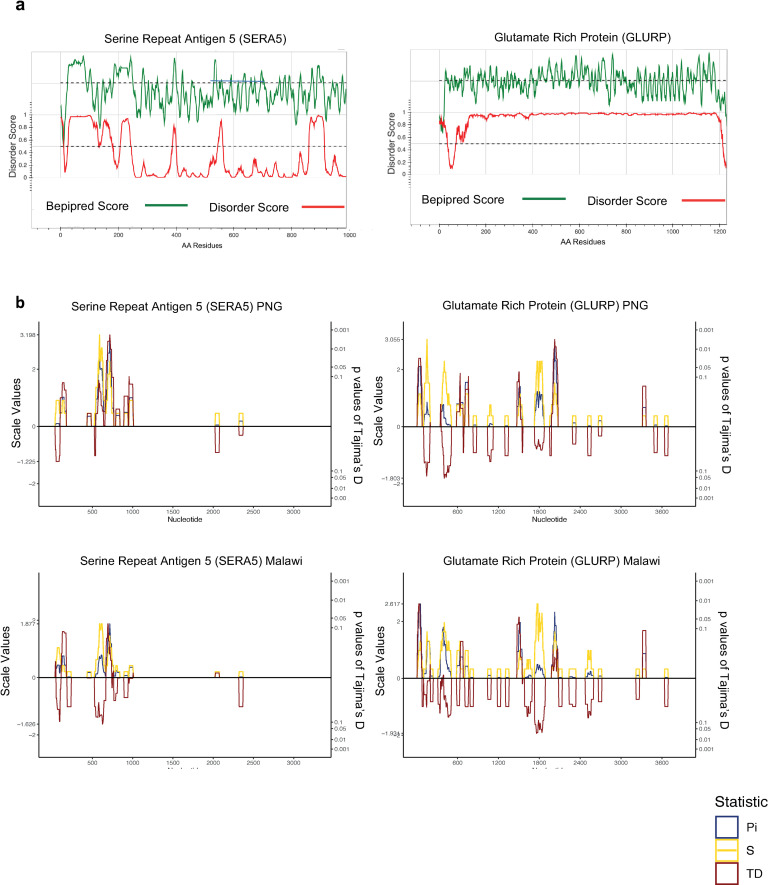
Investigation of the diversity of malaria parasite antigens can help prioritize and validate them as vaccine candidates and identify the most common variants for inclusion in vaccine formulations. Studies of vaccine candidates of the most virulent human malaria parasite, Plasmodium falciparum, have focused on a handful of well-known antigens, while several others have never been studied. Here we examine the global diversity and population structure of leading vaccine candidate antigens of P. falciparum using the MalariaGEN Pf3K (version 5.1) resource, comprising more than 2600 genomes from 15 malaria endemic countries. A stringent variant calling pipeline was used to extract high quality antigen gene 'haplotypes' from the global dataset and a new R-package named VaxPack was used to streamline population genetic analyses. In addition, a newly developed algorithm that enables spatial averaging of selection pressure on 3D protein structures was applied to the dataset. We analysed the genes encoding 23 leading and novel candidate malaria vaccine antigens including csp, trap, eba175, ama1, rh5, and CelTOS. Our analysis shows that current malaria vaccine formulations are based on rare haplotypes and thus may have limited efficacy against natural parasite populations. High levels of diversity with evidence of balancing selection was detected for most of the erythrocytic and pre-erythrocytic antigens. Measures of natural selection were then mapped to 3D protein structures to predict targets of functional antibodies. For some antigens, geographical variation in the intensity and distribution of these signals on the 3D structure suggests adaptation to different human host or mosquito vector populations. This study provides an essential framework for the diversity of P. falciparum antigens to be considered in the design of the next generation of malaria vaccines.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Diversity and selection analyses identify transmission-blocking antigens as the optimal vaccine candidates in Plasmodium falciparum.EBioMedicine. 2024 Aug;106:105227. doi: 10.1016/j.ebiom.2024.105227. Epub 2024 Jul 16. EBioMedicine. 2024. PMID: 39018754 Free PMC article.
-
Contrasting population structures of the genes encoding ten leading vaccine-candidate antigens of the human malaria parasite, Plasmodium falciparum.PLoS One. 2009 Dec 30;4(12):e8497. doi: 10.1371/journal.pone.0008497. PLoS One. 2009. PMID: 20041125 Free PMC article.
-
Global diversity of the gene encoding the Pfs25 protein-a Plasmodium falciparum transmission-blocking vaccine candidate.Parasit Vectors. 2021 Nov 8;14(1):571. doi: 10.1186/s13071-021-05078-6. Parasit Vectors. 2021. PMID: 34749796 Free PMC article.
-
Current Challenges in the Identification of Pre-Erythrocytic Malaria Vaccine Candidate Antigens.Front Immunol. 2020 Feb 21;11:190. doi: 10.3389/fimmu.2020.00190. eCollection 2020. Front Immunol. 2020. PMID: 32153565 Free PMC article. Review.
-
Antigens for pre-erythrocytic malaria vaccines: building on success.Parasite Immunol. 2009 Sep;31(9):539-46. doi: 10.1111/j.1365-3024.2009.01139.x. Parasite Immunol. 2009. PMID: 19691557 Free PMC article. Review.
Cited by
-
Computational Clues of Immunogenic Hotspots in Plasmodium falciparum Erythrocytic Stage Vaccine Candidate Antigens: In Silico Approach.Biomed Res Int. 2022 Oct 13;2022:5886687. doi: 10.1155/2022/5886687. eCollection 2022. Biomed Res Int. 2022. PMID: 36277884 Free PMC article.
-
High Prevalence of Polyclonal Plasmodium falciparum Infections and Association with Poor IgG Antibody Responses in a Hyper-Endemic Area in Cameroon.Trop Med Infect Dis. 2023 Jul 29;8(8):390. doi: 10.3390/tropicalmed8080390. Trop Med Infect Dis. 2023. PMID: 37624328 Free PMC article.
-
Diversity and selection analyses identify transmission-blocking antigens as the optimal vaccine candidates in Plasmodium falciparum.EBioMedicine. 2024 Aug;106:105227. doi: 10.1016/j.ebiom.2024.105227. Epub 2024 Jul 16. EBioMedicine. 2024. PMID: 39018754 Free PMC article.
-
Ancient diversity in host-parasite interaction genes in a model parasitic nematode.Nat Commun. 2023 Nov 27;14(1):7776. doi: 10.1038/s41467-023-43556-w. Nat Commun. 2023. PMID: 38012132 Free PMC article.
-
Population Genomics of Plasmodium malariae from Four African Countries.medRxiv [Preprint]. 2024 Sep 9:2024.09.07.24313132. doi: 10.1101/2024.09.07.24313132. medRxiv. 2024. PMID: 39314932 Free PMC article. Preprint.
References
-
- Mobegi VA, Duffy CW, Amambua-Ngwa A, Loua KM, Laman E, Nwakanma DC, et al.. Genome-Wide Analysis of Selection on the Malaria Parasite Plasmodium falciparum in West African Populations of Differing Infection Endemicity. Mol Biol Evol. 2014;31: 1490–1499. doi: 10.1093/molbev/msu106 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
