

Staphylococcus aureus and Neutrophil Extracellular Traps: The Master Manipulator Meets Its Match in Immunothrombosis
- PMID: 35109674
- PMCID: PMC8860219
- DOI: 10.1161/ATVBAHA.121.316930
Staphylococcus aureus and Neutrophil Extracellular Traps: The Master Manipulator Meets Its Match in Immunothrombosis
Abstract
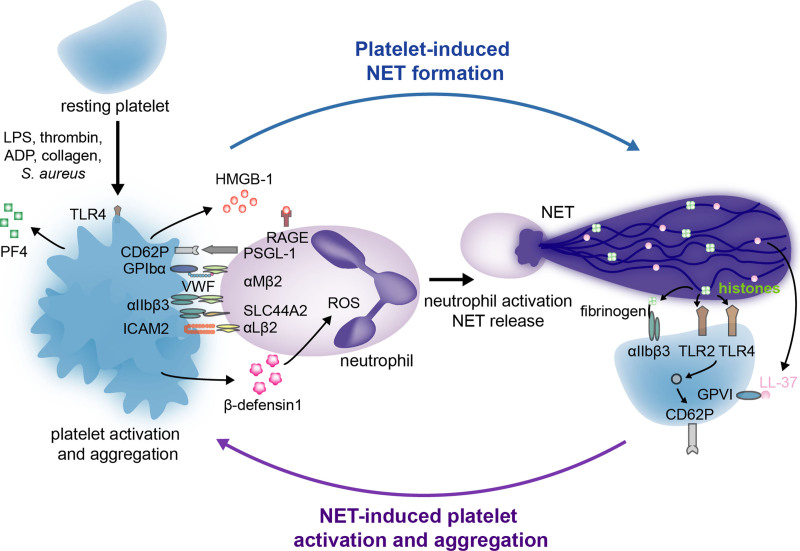
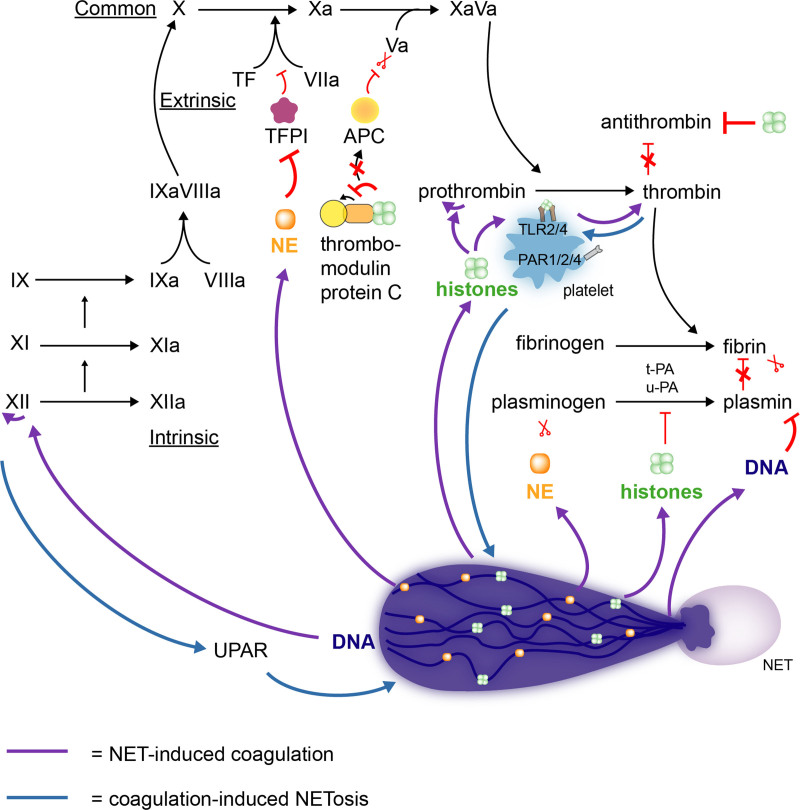
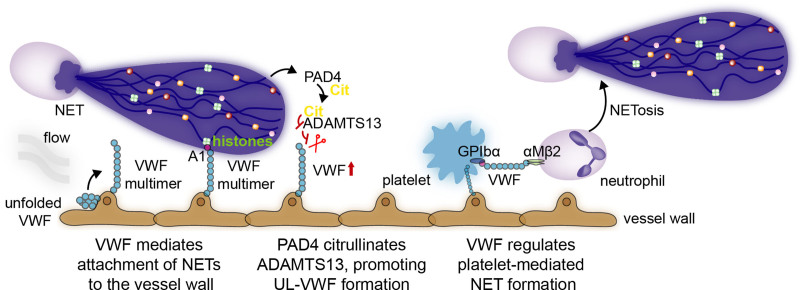
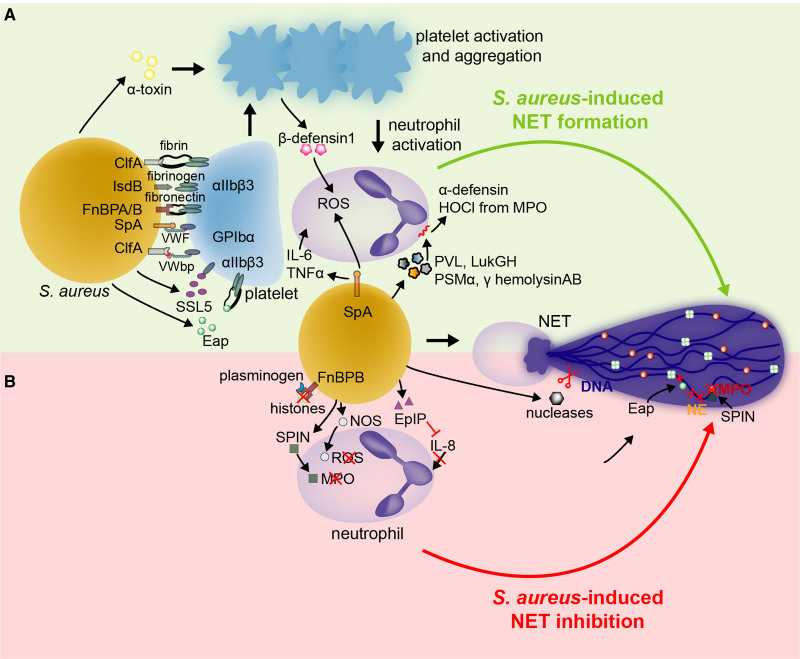
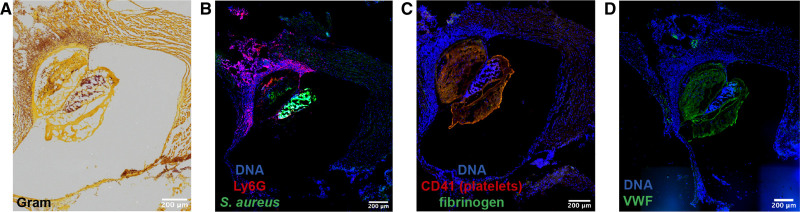
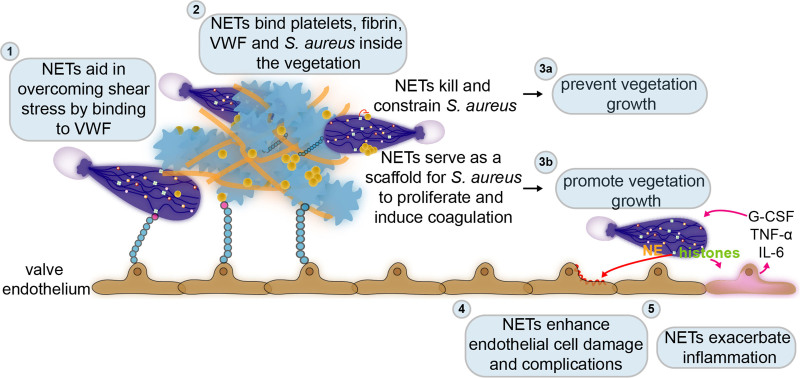
Over the past 10 years, neutrophil extracellular traps (NETs) have become widely accepted as an integral player in immunothrombosis, due to their complex interplay with both pathogens and components of the coagulation system. While the release of NETs is an attempt by neutrophils to trap pathogens and constrain infections, NETs can have bystander effects on the host by inducing uncontrolled thrombosis, inflammation, and tissue damage. From an evolutionary perspective, pathogens have adapted to bypass the host innate immune response. Staphylococcus aureus (S. aureus), in particular, proficiently overcomes NET formation using several virulence factors. Here we review mechanisms of NET formation and how these are intertwined with platelet activation, the release of endothelial von Willebrand factor, and the activation of the coagulation system. We discuss the unique ability of S. aureus to modulate NET formation and alter released NETs, which helps S. aureus to escape from the host's defense mechanisms. We then discuss how platelets and the coagulation system could play a role in NET formation in S. aureus-induced infective endocarditis, and we explain how targeting these complex cellular interactions could reveal novel therapies to treat this disease and other immunothrombotic disorders.
Keywords: Staphylococcus; endocarditis; extracellular trap; neutrophils; platelet activation; virulence factors.
Figures
References
-
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385 - PubMed
-
- Tsan MF. Phorbol myristate acetate induced neutrophil autotoxicity. J Cell Physiol. 1980;105:327–334. doi: 10.1002/jcp.1041050215 - PubMed
-
- Tsan MF, Denison RC. Phorbol myristate acetate-induced neutrophil autotoxicity. A comparison with H2O2 toxicity. Inflammation. 1980;4:371–380. doi: 10.1007/BF00916048 - PubMed
-
- Wooldridge LC. XVI. The relation of the white blood corpuscles to the coagulation of the blood. Proc R Soc Lond. 1997;32:413–418.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
