

Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice
- PMID: 35112666
- PMCID: PMC8846590
- DOI: 10.7554/eLife.74419
Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice
Abstract
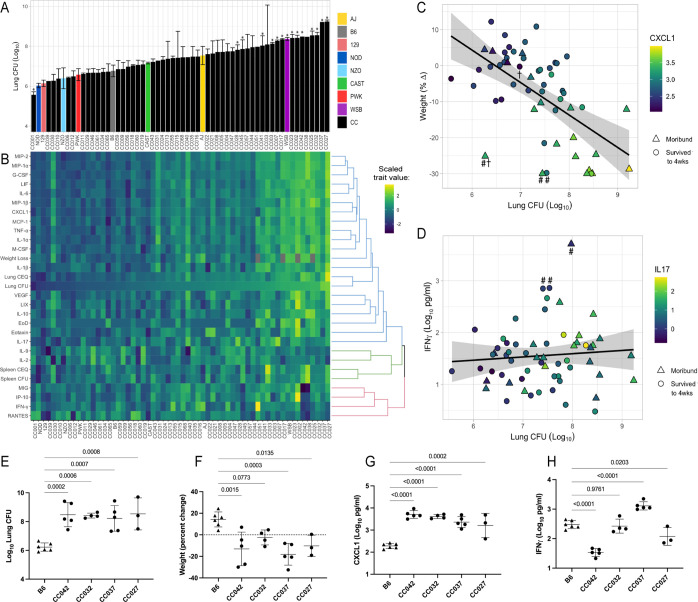
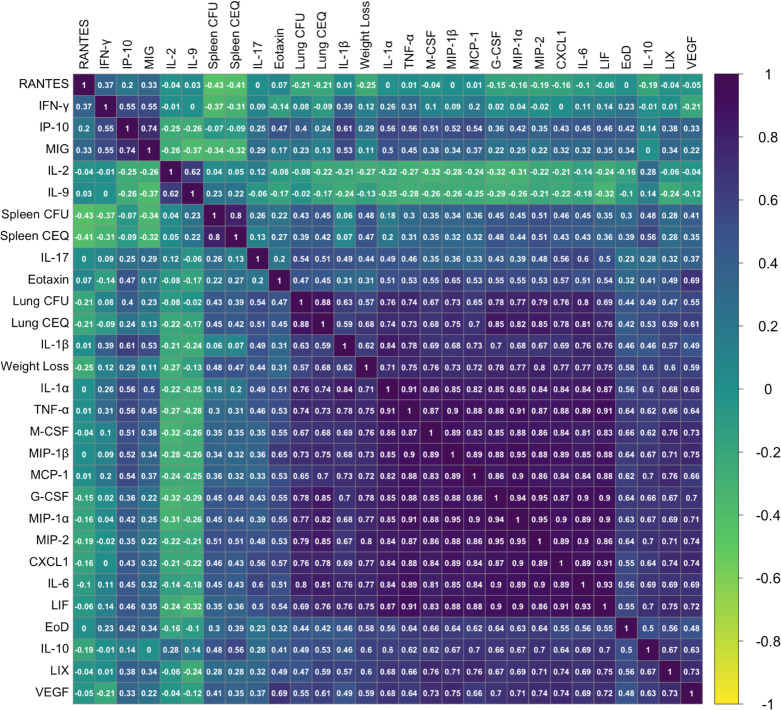
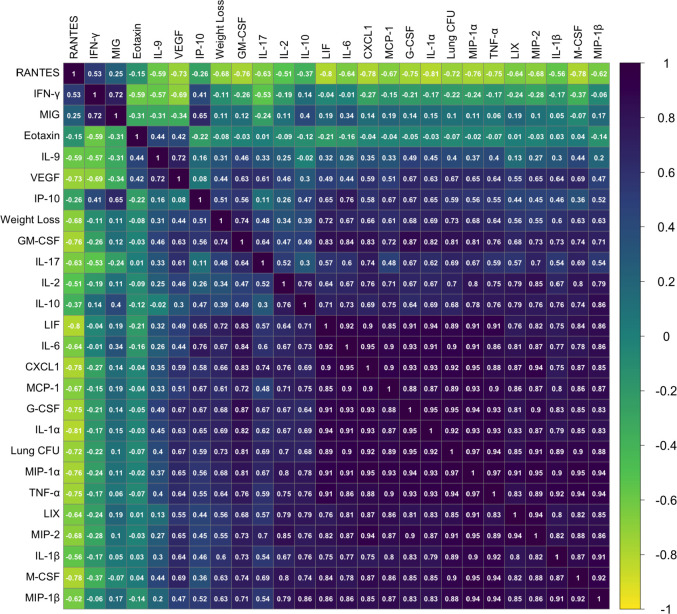
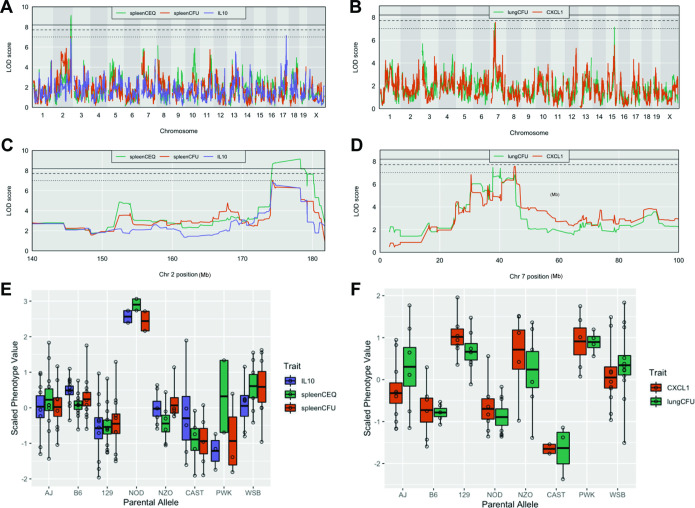
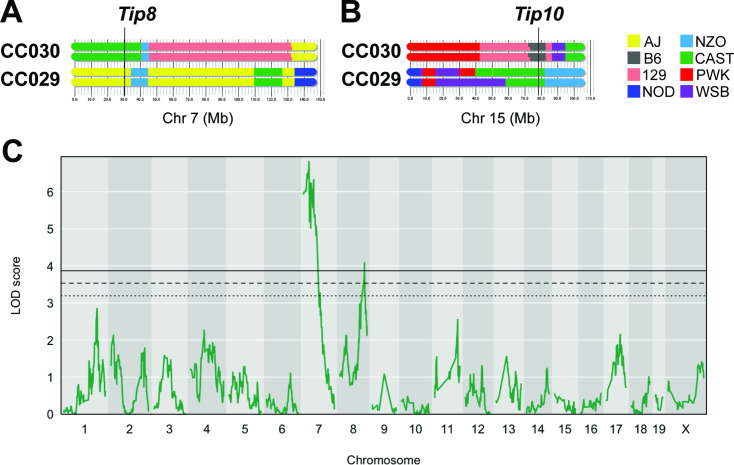
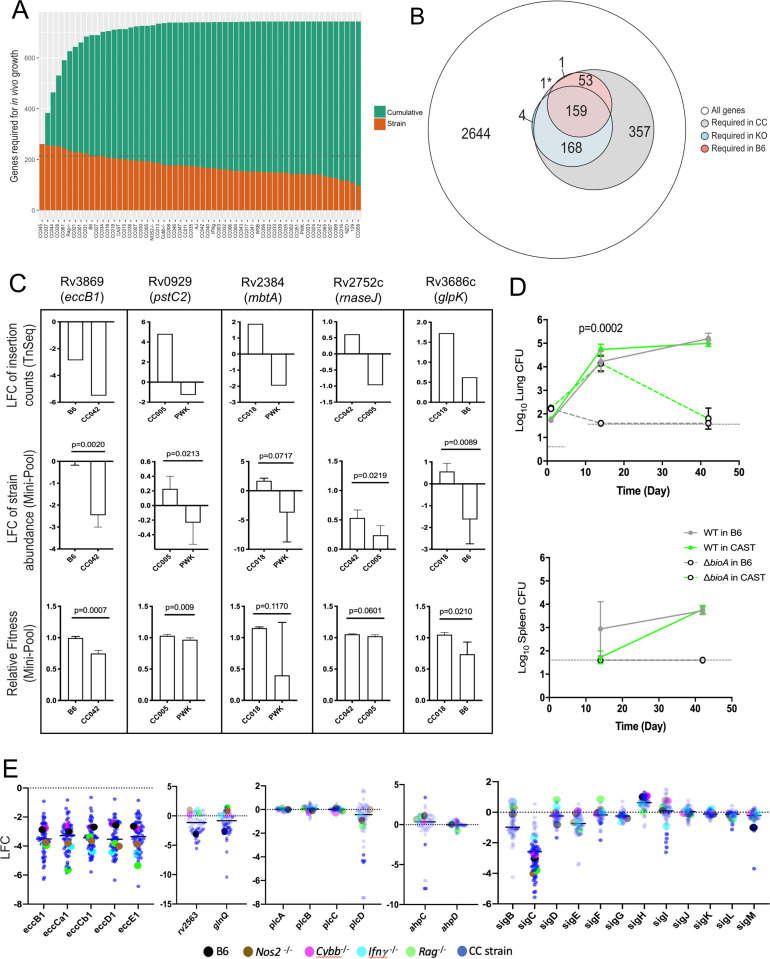
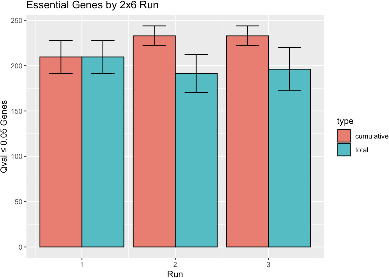
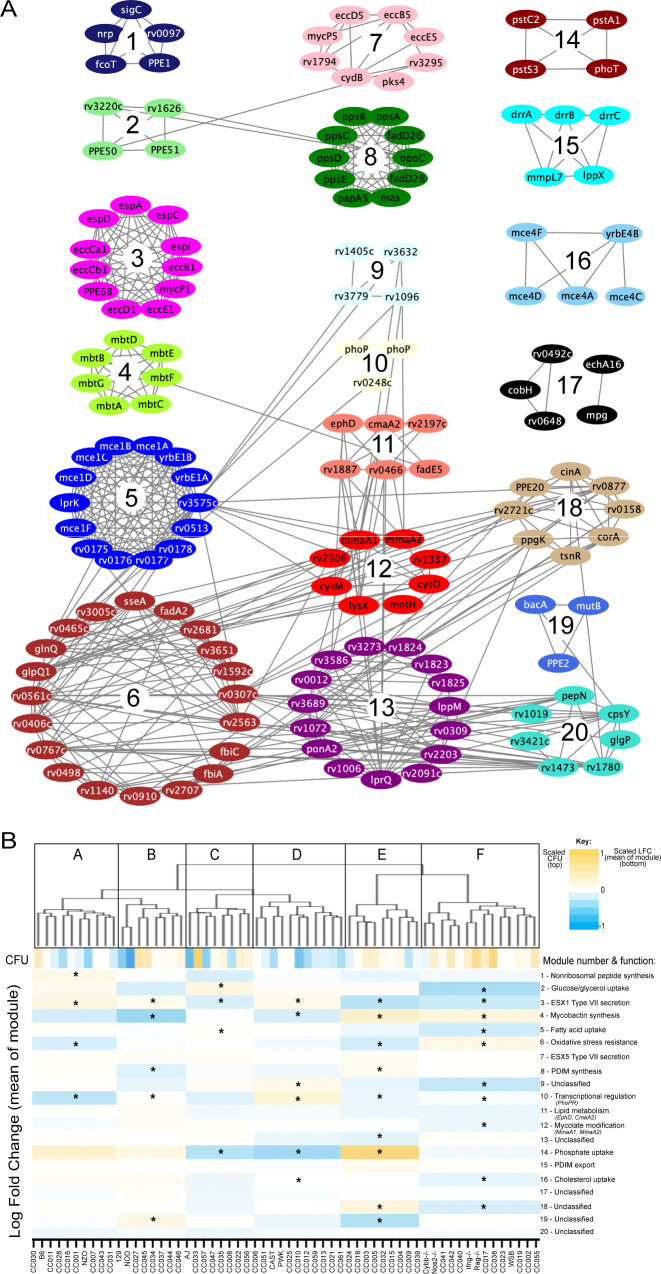
The outcome of an encounter with Mycobacterium tuberculosis (Mtb) depends on the pathogen's ability to adapt to the variable immune pressures exerted by the host. Understanding this interplay has proven difficult, largely because experimentally tractable animal models do not recapitulate the heterogeneity of tuberculosis disease. We leveraged the genetically diverse Collaborative Cross (CC) mouse panel in conjunction with a library of Mtb mutants to create a resource for associating bacterial genetic requirements with host genetics and immunity. We report that CC strains vary dramatically in their susceptibility to infection and produce qualitatively distinct immune states. Global analysis of Mtb transposon mutant fitness (TnSeq) across the CC panel revealed that many virulence pathways are only required in specific host microenvironments, identifying a large fraction of the pathogen's genome that has been maintained to ensure fitness in a diverse population. Both immunological and bacterial traits can be associated with genetic variants distributed across the mouse genome, making the CC a unique population for identifying specific host-pathogen genetic interactions that influence pathogenesis.
Keywords: TnSeq; collaborative cross; genetics; genomics; host-pathogen interactions; infectious disease; microbiology; mouse; mouse models; systems genetics; tuberculosis.
© 2022, Smith et al.
Conflict of interest statement
CS, RB, MP, BM, JL, SP, HL, MK, MB, AO, KM, KP, FB, CR, RM, BH, CL, GS, PH, TB, SE, DS, FP, MF, TI, CS No competing interests declared
Figures
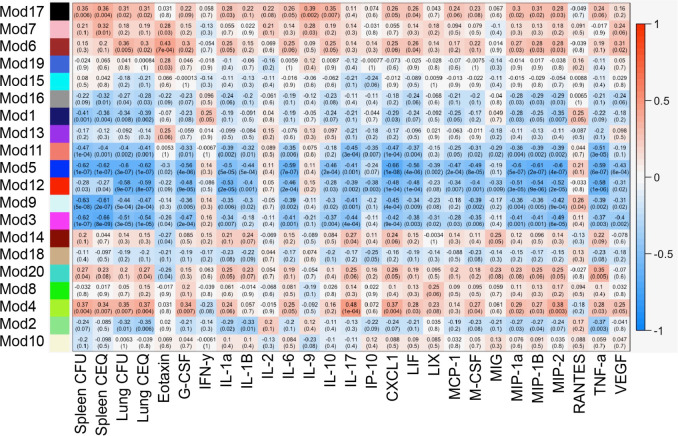
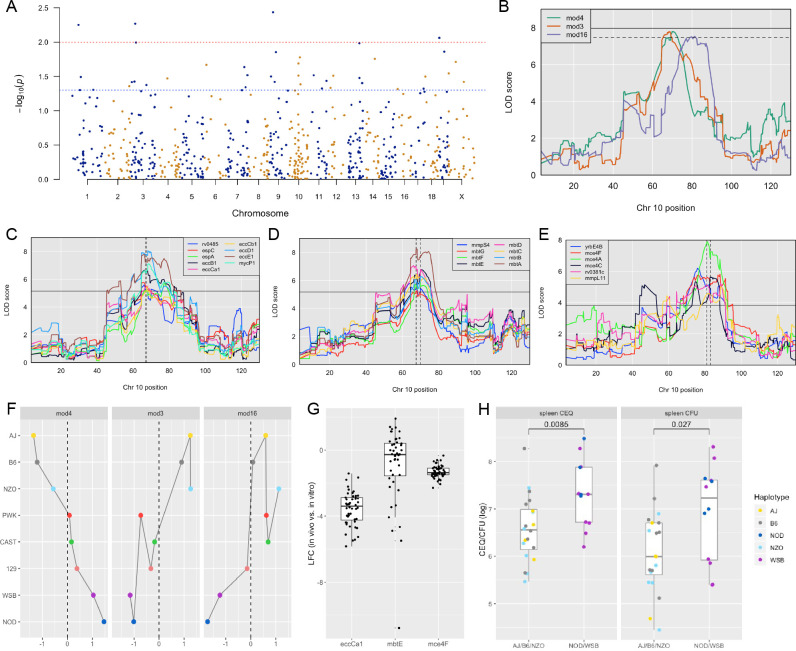
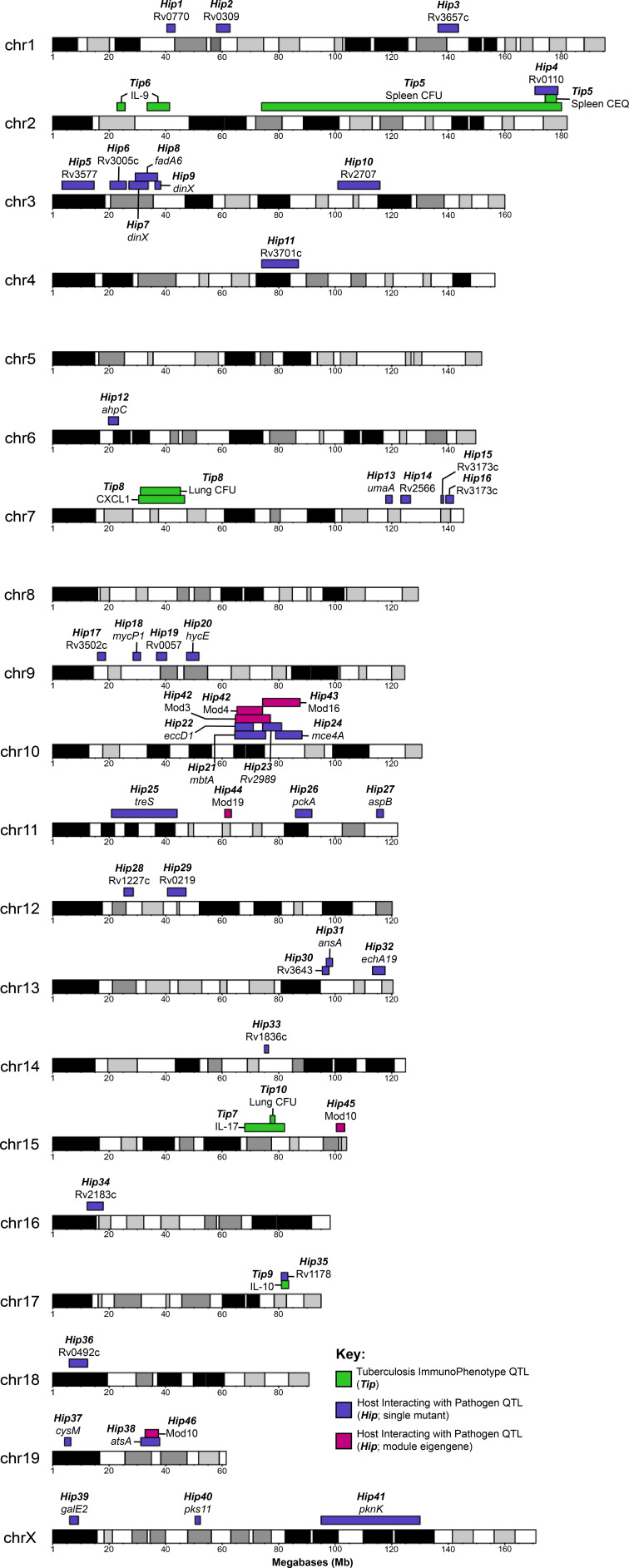
Comment in
-
The Collaborative Cross: a model for genetic variation in TB.Lab Anim (NY). 2022 Apr;51(4):98. doi: 10.1038/s41684-022-00953-w. Lab Anim (NY). 2022. PMID: 35361983 No abstract available.
References
-
- Abel L, Fellay J, Haas DW, Schurr E, Srikrishna G, Urbanowski M, Chaturvedi N, Srinivasan S, Johnson DH, Bishai WR. Genetics of human susceptibility to active and latent tuberculosis: present knowledge and future perspectives. The Lancet. Infectious Diseases. 2018;18:e64–e75. doi: 10.1016/S1473-3099(17)30623-0. - DOI - PMC - PubMed
-
- Ahmed M, Thirunavukkarasu S, Rosa BA, Thomas KA, Das S, Rangel-Moreno J, Lu L, Mehra S, Mbandi SK, Thackray LB, Diamond MS, Murphy KM, Means T, Martin J, Kaushal D, Scriba TJ, Mitreva M, Khader SA. Immune correlates of tuberculosis disease and risk translate across species. Science Translational Medicine. 2020;12:eaay0233. doi: 10.1126/scitranslmed.aay0233. - DOI - PMC - PubMed
-
- Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Döffinger R, Bernaudin F, Jeppsson O, Gollob JA, Meinl E, Segal AW, Fischer A, Kumararatne D, Casanova JL. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science (New York, N.Y.) 1998;280:1432–1435. doi: 10.1126/science.280.5368.1432. - DOI - PubMed
-
- Amaral EP, Riteau N, Moayeri M, Maier N, Mayer-Barber KD, Pereira RM, Lage SL, Kubler A, Bishai WR, D’Império-Lima MR, Sher A, Andrade BB. Lysosomal Cathepsin Release Is Required for NLRP3-Inflammasome Activation by Mycobacterium tuberculosis in Infected Macrophages. Frontiers in Immunology. 2018;9:1427. doi: 10.3389/fimmu.2018.01427. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
