

CCL17 Aggravates Myocardial Injury by Suppressing Recruitment of Regulatory T Cells
- PMID: 35113652
- PMCID: PMC8957788
- DOI: 10.1161/CIRCULATIONAHA.121.055888
CCL17 Aggravates Myocardial Injury by Suppressing Recruitment of Regulatory T Cells
Abstract
Background: Recent studies have established that CCR2 (C-C chemokine receptor type 2) marks proinflammatory subsets of monocytes, macrophages, and dendritic cells that contribute to adverse left ventricle (LV) remodeling and heart failure progression. Elucidation of the effector mechanisms that mediate adverse effects of CCR2+ monocytes, macrophages, and dendritic cells will yield important insights into therapeutic strategies to suppress myocardial inflammation.
Methods: We used mouse models of reperfused myocardial infarction, angiotensin II and phenylephrine infusion, and diphtheria toxin cardiomyocyte ablation to investigate CCL17 (C-C chemokine ligand 17). We used Ccl17 knockout mice, flow cytometry, RNA sequencing, biochemical assays, cell trafficking studies, and in vivo cell depletion to identify the cell types that generate CCL17, define signaling pathways that controlled its expression, delineate the functional importance of CCL17 in adverse LV remodeling and heart failure progression, and determine the mechanistic basis by which CCL17 exerts its effects.
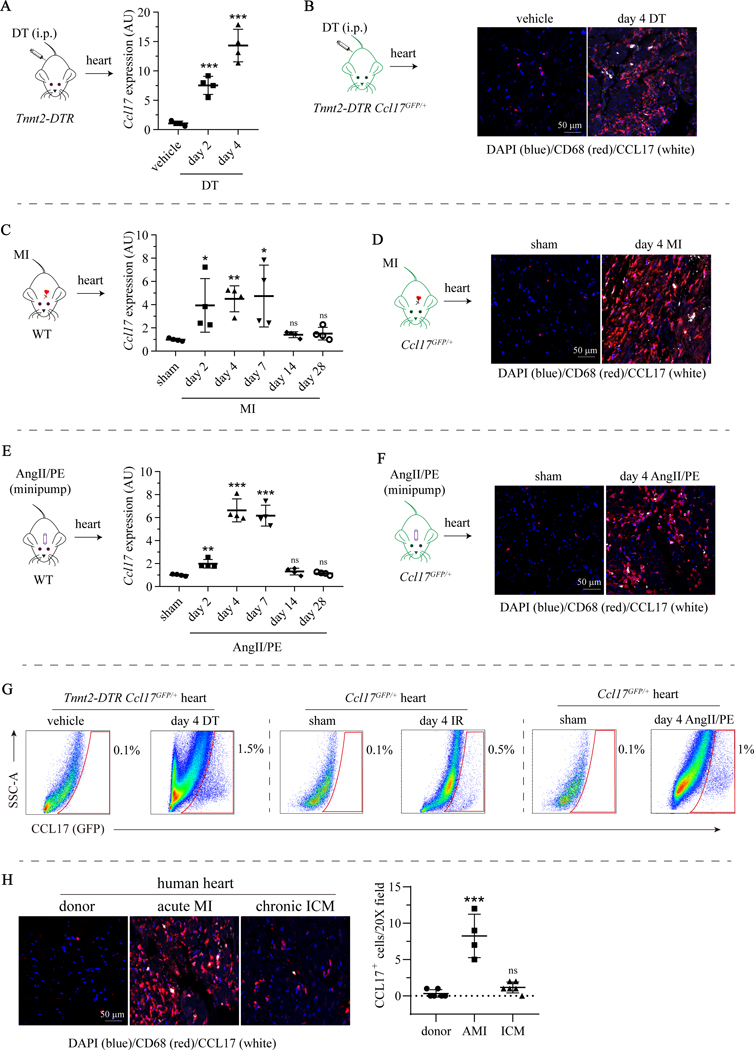
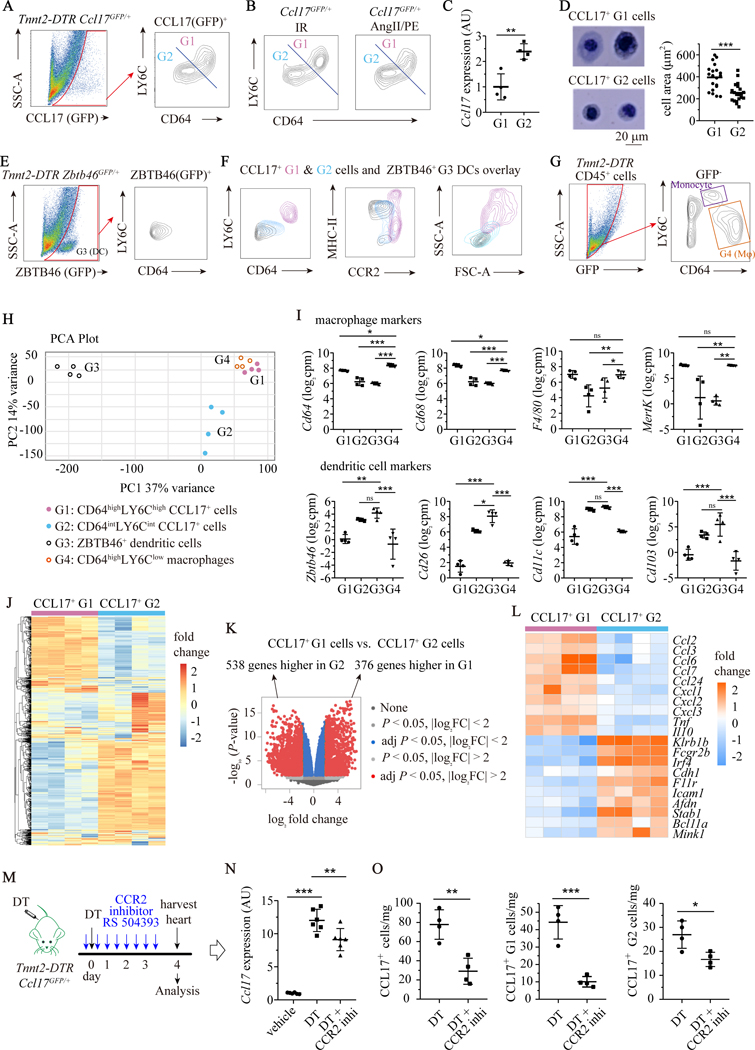
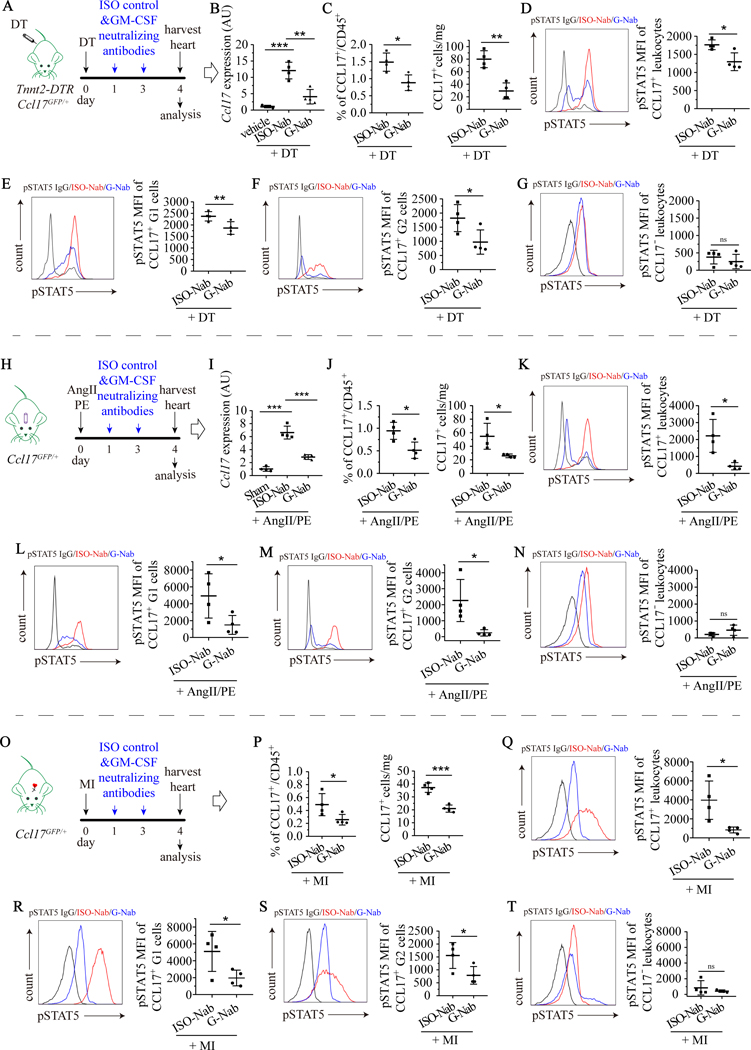
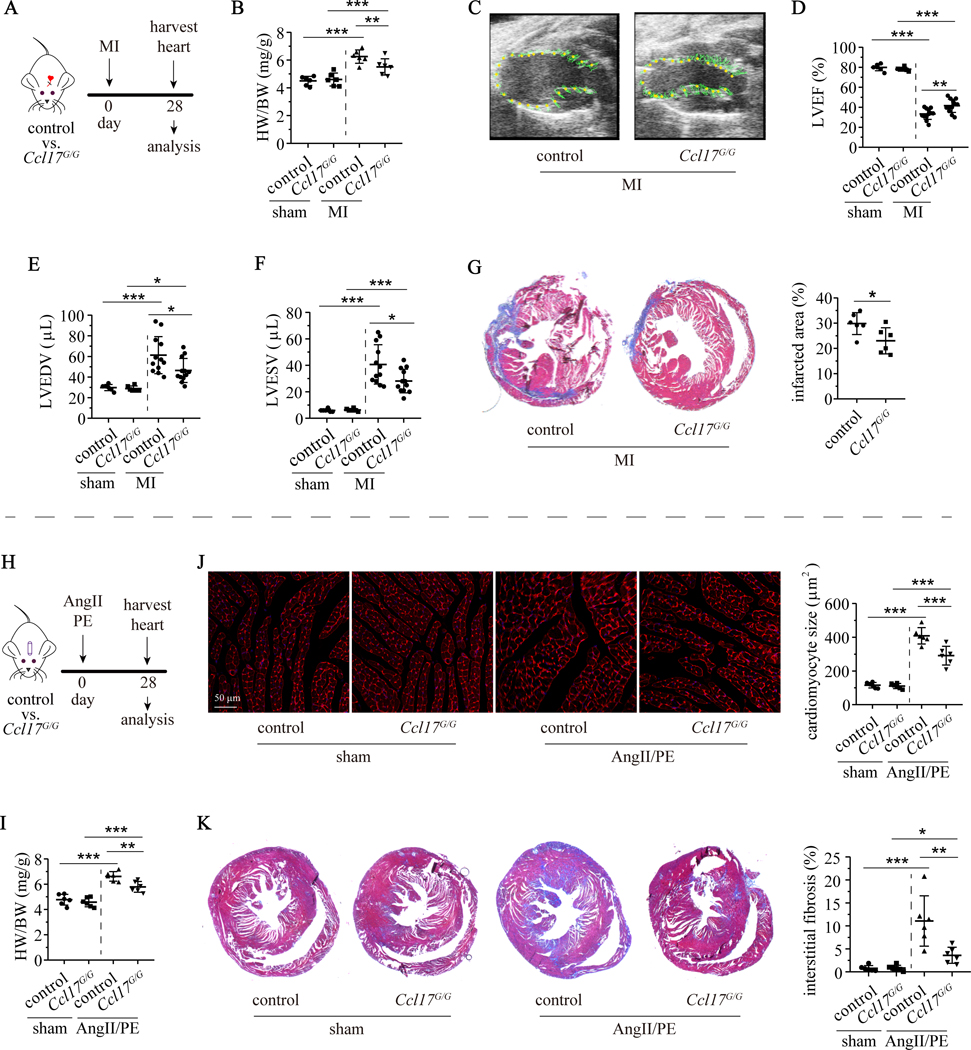
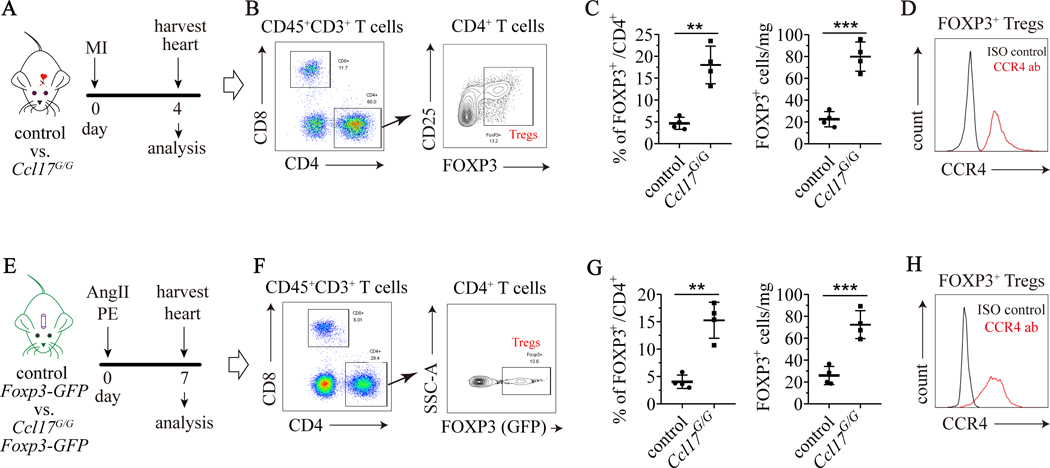
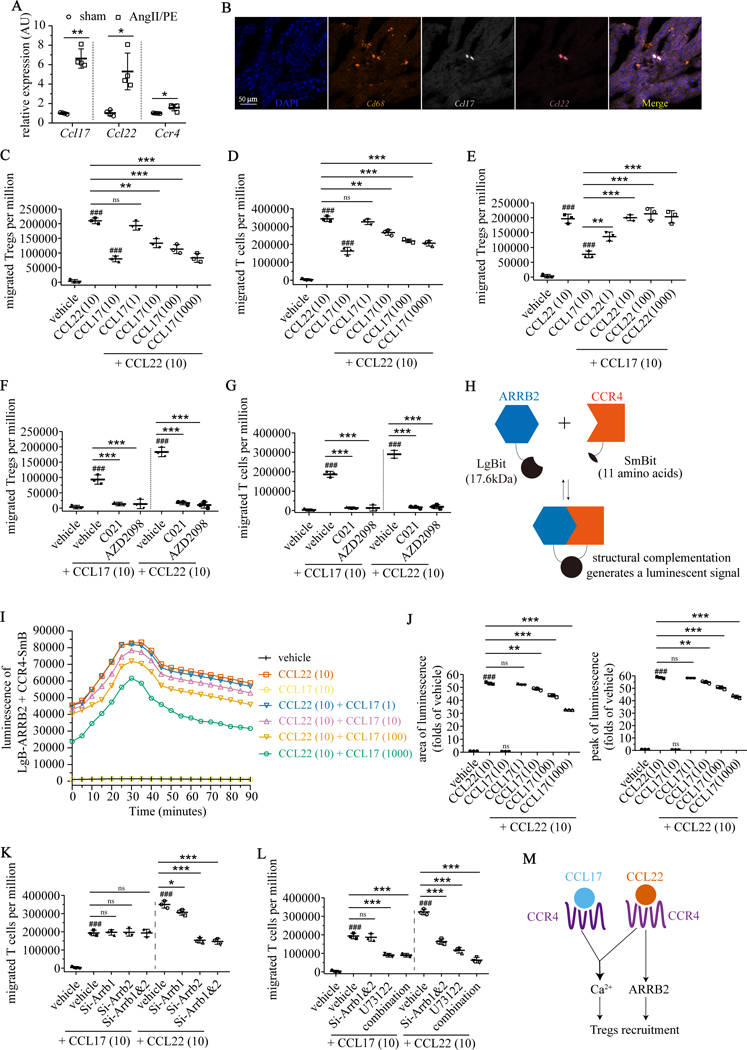
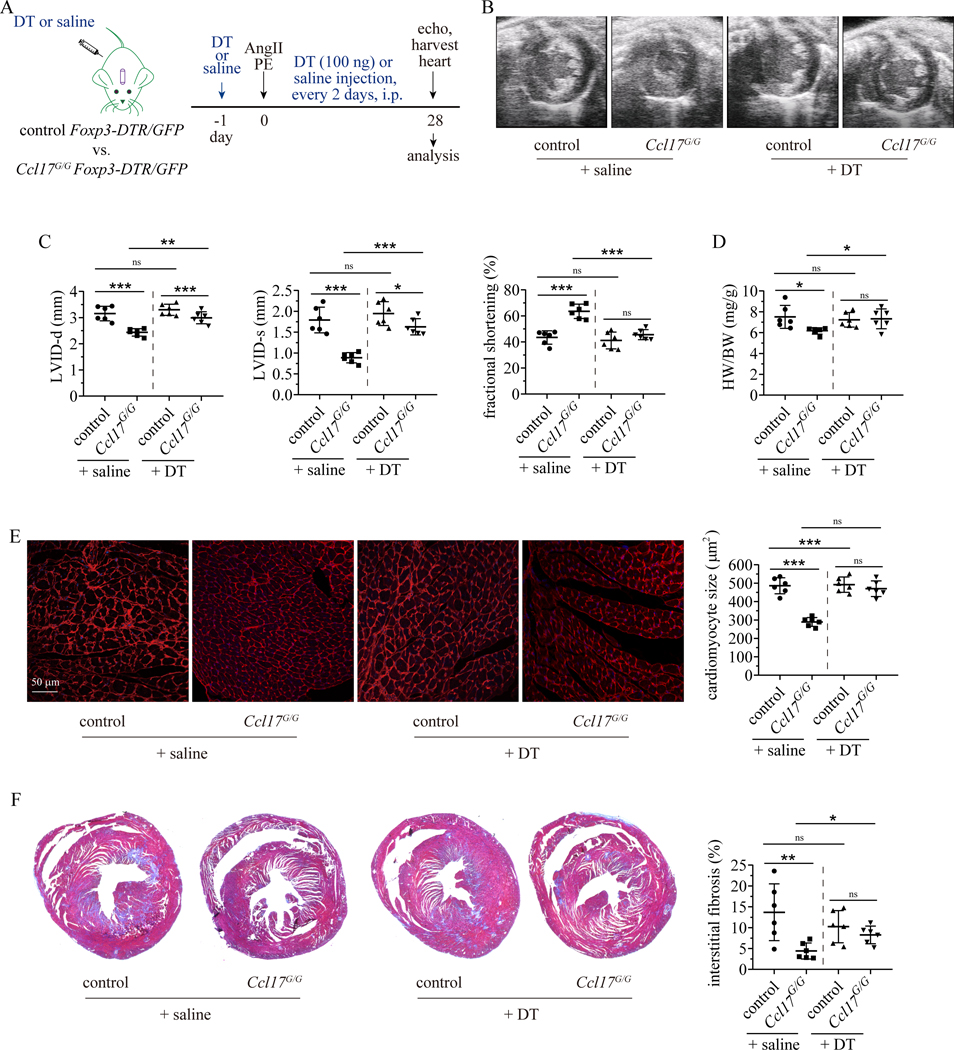
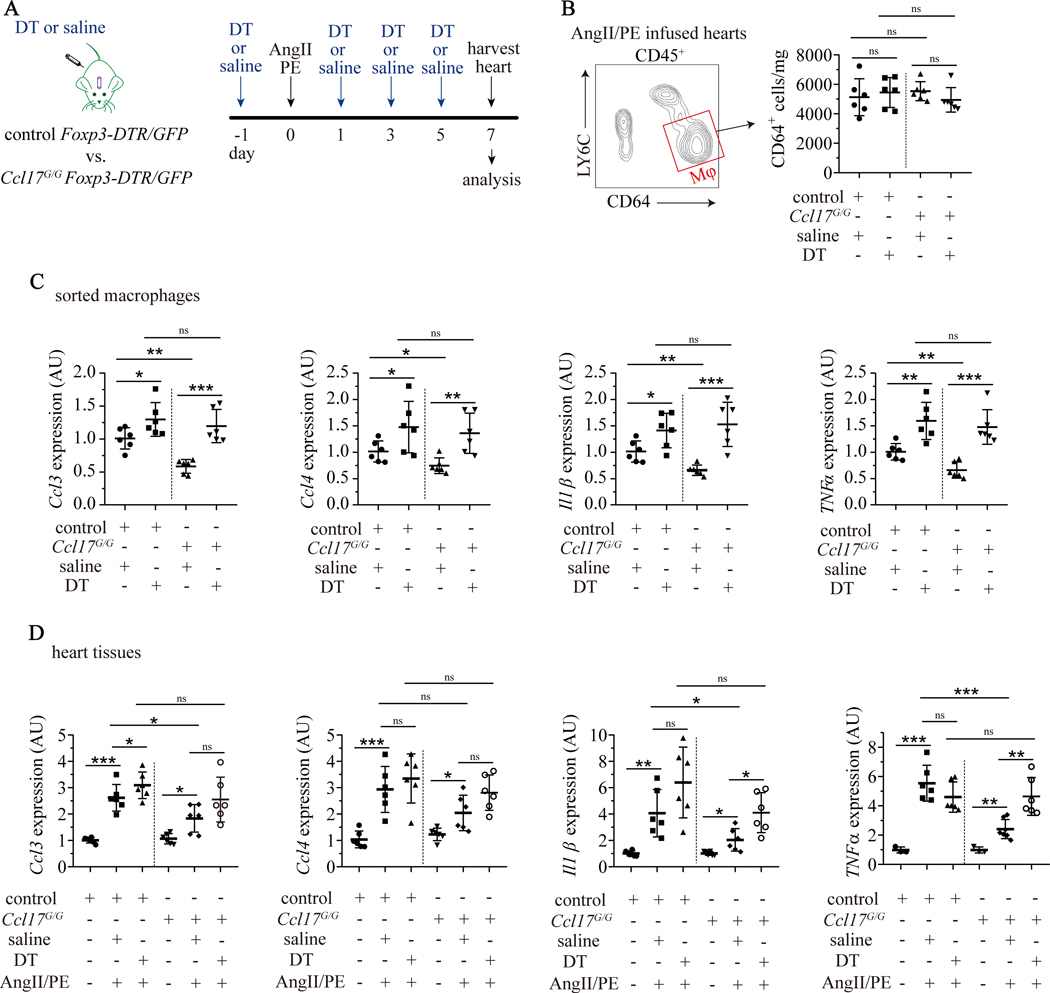
Results: We demonstrated that CCL17 is expressed in CCR2+ macrophages and cluster of differentiation 11b+ conventional dendritic cells after myocardial infarction, angiotensin II and phenylephrine infusion, and diphtheria toxin cardiomyocyte ablation. We clarified the transcriptional signature of CCL17+ macrophages and dendritic cells and identified granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling as a key regulator of CCL17 expression through cooperative activation of STAT5 (signal transducer and activator of transcription 5) and canonical NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) signaling. Ccl17 deletion resulted in reduced LV remodeling, decreased myocardial fibrosis and cardiomyocyte hypertrophy, and improved LV systolic function after myocardial infarction and angiotensin II and phenylephrine infusion. We observed increased abundance of regulatory T cells (Tregs) in the myocardium of injured Ccl17 knockout mice. CCL17 inhibited Treg recruitment through biased activation of CCR4. CCL17 activated Gq signaling and CCL22 (C-C chemokine ligand 22) activated both Gq and ARRB (β-arrestin) signaling downstream of CCR4. CCL17 competitively inhibited CCL22 stimulated ARRB signaling and Treg migration. We provide evidence that Tregs mediated the protective effects of Ccl17 deletion on myocardial inflammation and adverse LV remodeling.
Conclusions: These findings identify CCL17 as a proinflammatory mediator of CCR2+ macrophages and dendritic cells and suggest that inhibition of CCL17 may serve as an effective strategy to promote Treg recruitment and suppress myocardial inflammation.
Keywords: T-lymphocytes, regulatory; chemokine CCL17; dendritic cells; inflammation; macrophages; monocytes.
Figures
References
-
- Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, Von Schlippenbach J, Skurk C, Steendijk P, Riad A. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circulation: Heart Failure. 2011;4:44–52. - PubMed
-
- Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM. Inflammation in myocardial diseases. Circulation research. 2012;110:126–144. - PubMed
-
- Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology. 2012;122:23–35. - PubMed
-
- Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nature Reviews Cardiology. 2020;17:269–285. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
