

Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort
- PMID: 35115689
- PMCID: PMC9883041
- DOI: 10.1038/s41588-021-00991-z
Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort
Erratum in
-
Author Correction: Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort.Nat Genet. 2024 Mar;56(3):554. doi: 10.1038/s41588-024-01693-y. Nat Genet. 2024. PMID: 38424462 No abstract available.
Abstract
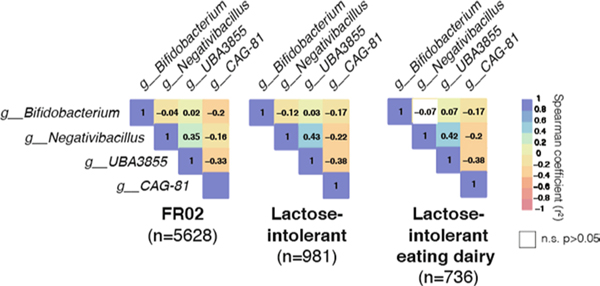
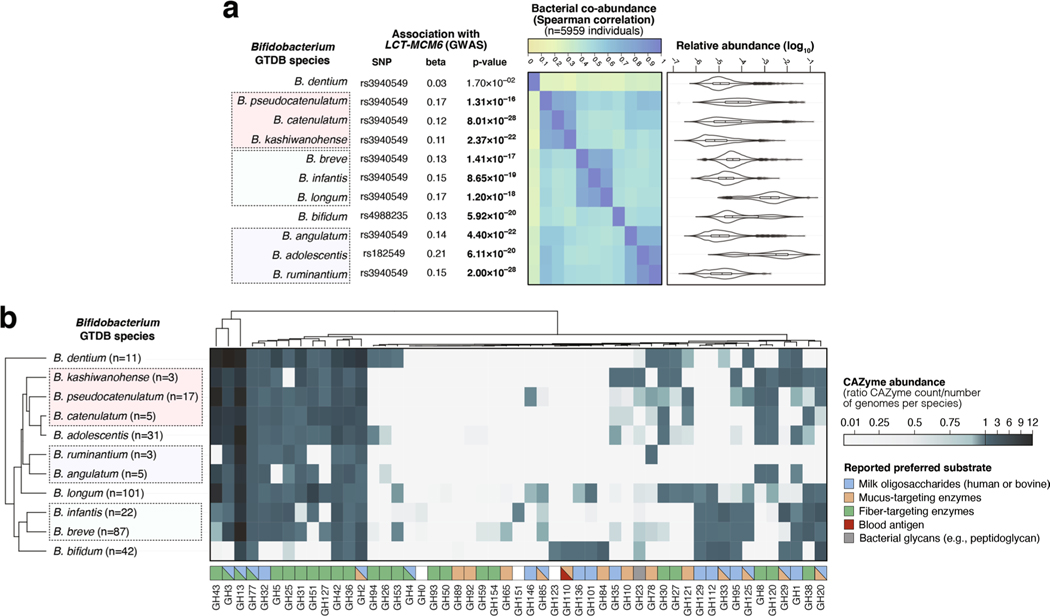
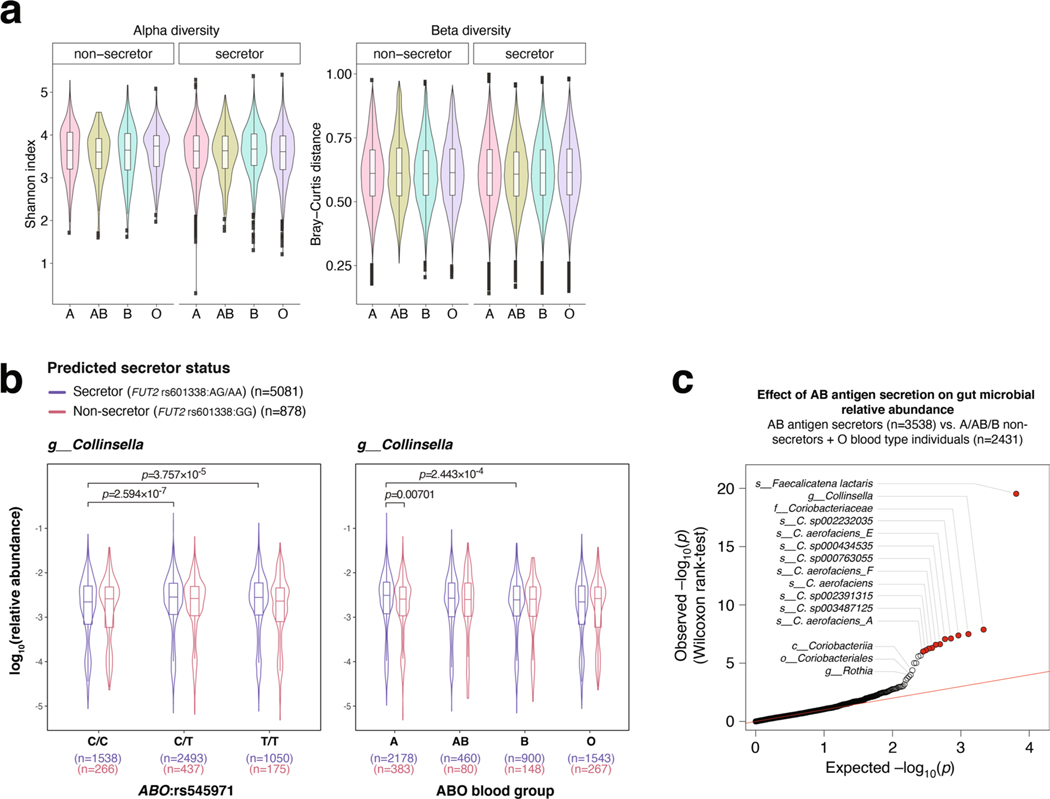
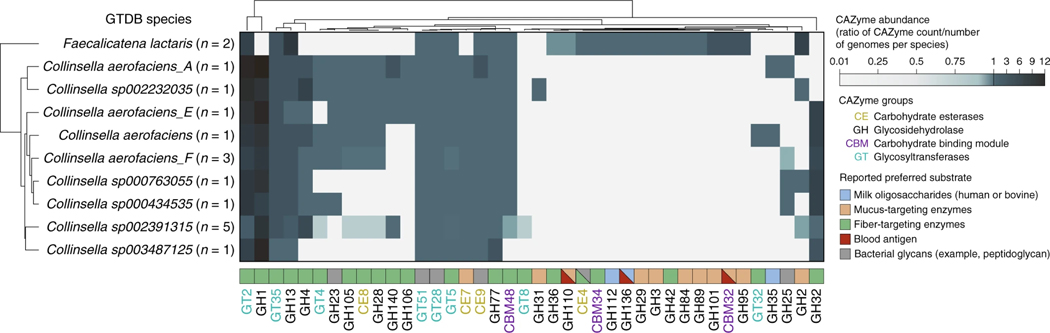
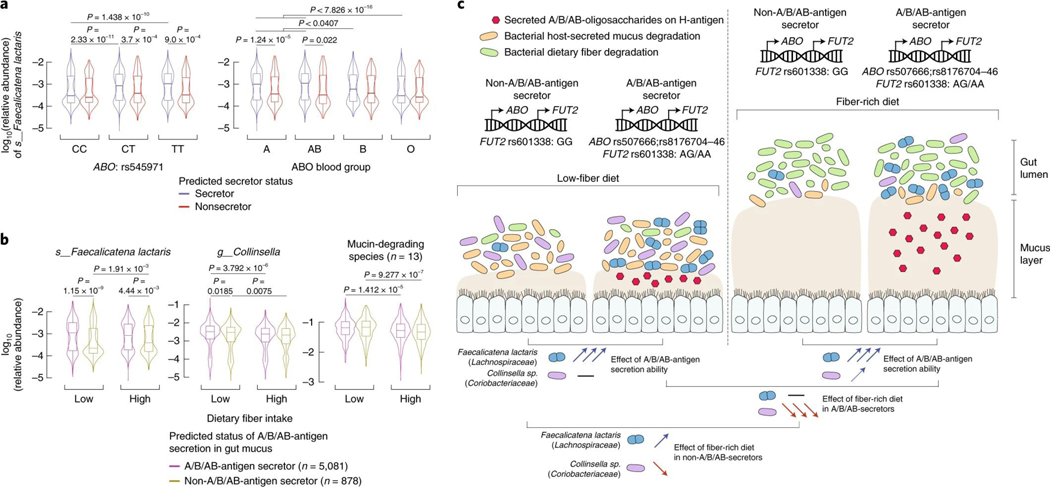
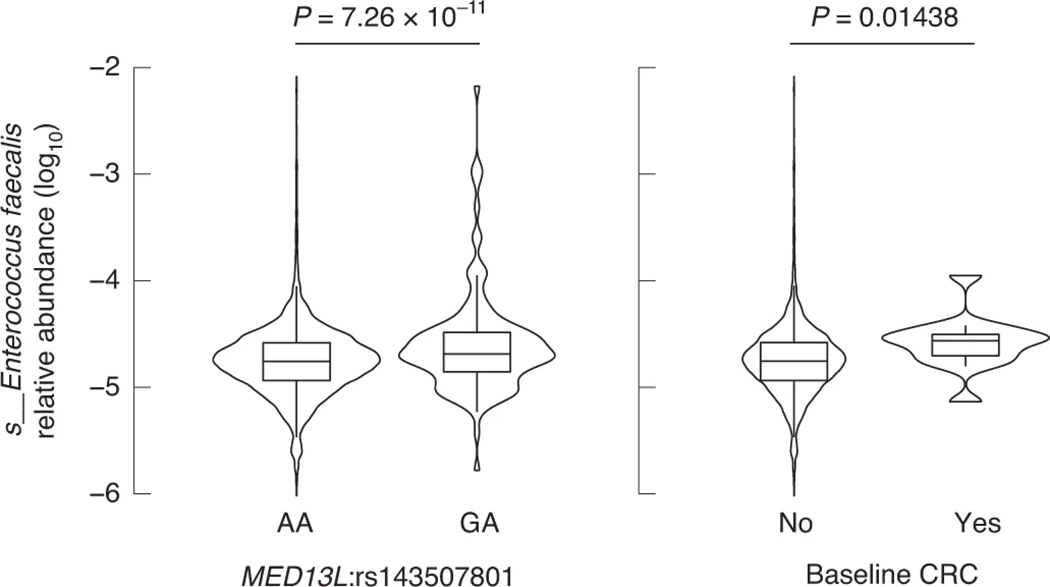
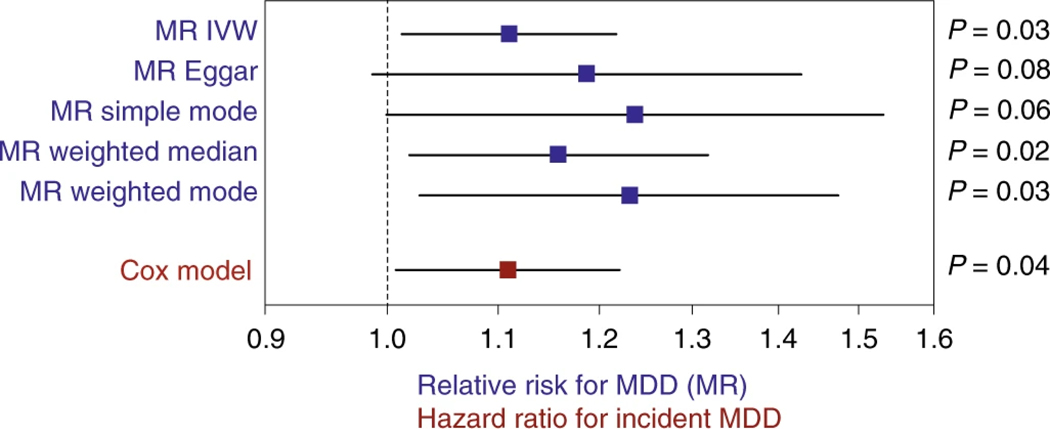
Human genetic variation affects the gut microbiota through a complex combination of environmental and host factors. Here we characterize genetic variations associated with microbial abundances in a single large-scale population-based cohort of 5,959 genotyped individuals with matched gut microbial metagenomes, and dietary and health records (prevalent and follow-up). We identified 567 independent SNP-taxon associations. Variants at the LCT locus associated with Bifidobacterium and other taxa, but they differed according to dairy intake. Furthermore, levels of Faecalicatena lactaris associated with ABO, and suggested preferential utilization of secreted blood antigens as energy source in the gut. Enterococcus faecalis levels associated with variants in the MED13L locus, which has been linked to colorectal cancer. Mendelian randomization analysis indicated a potential causal effect of Morganella on major depressive disorder, consistent with observational incident disease analysis. Overall, we identify and characterize the intricate nature of host-microbiota interactions and their association with disease.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
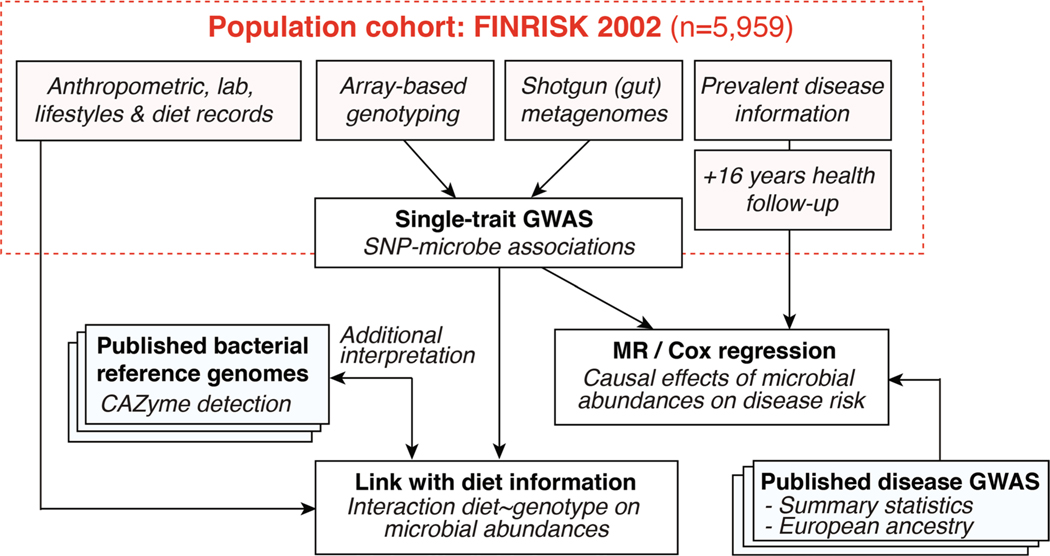
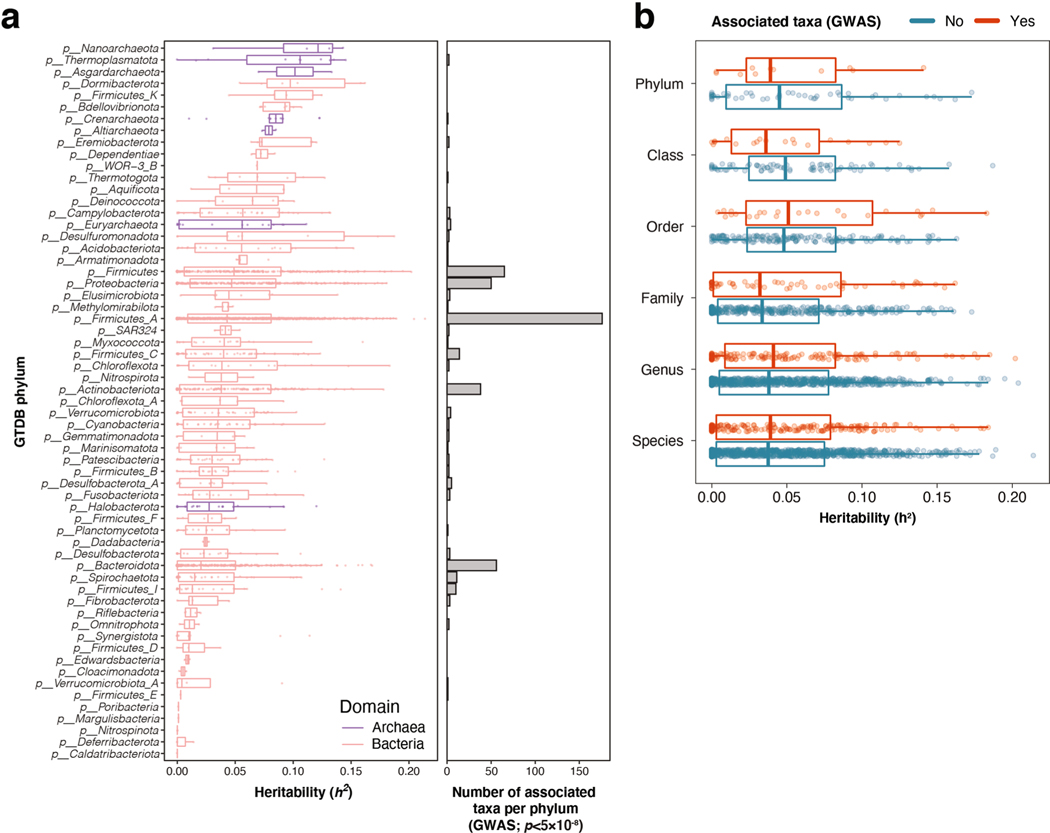
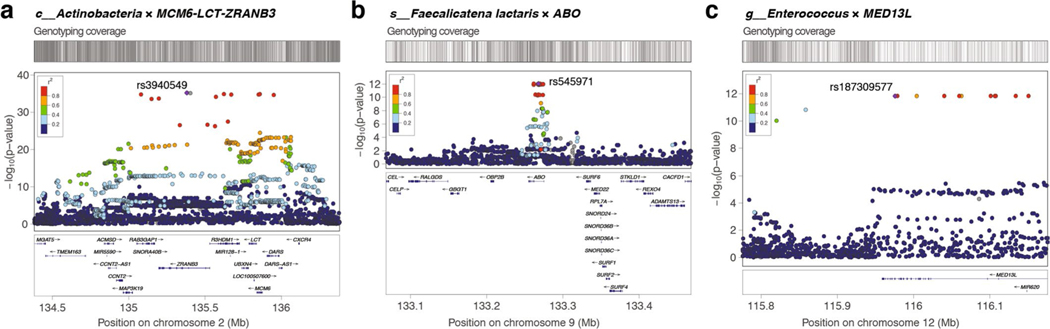
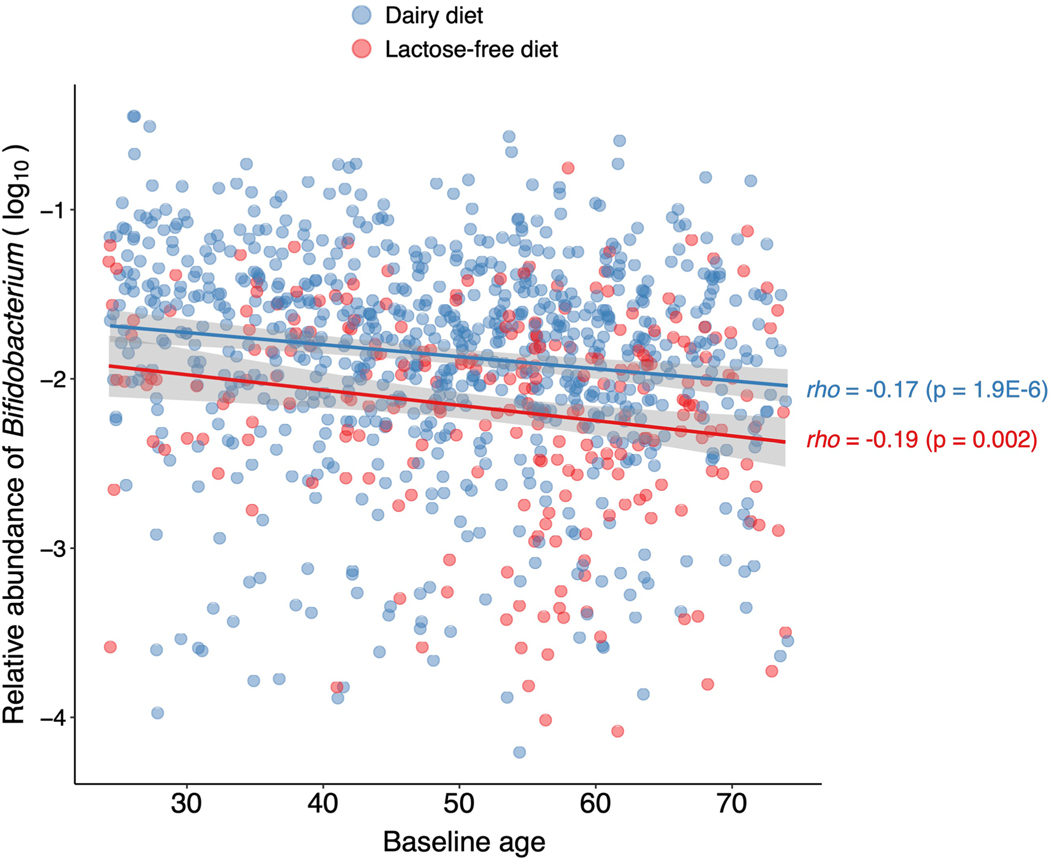
References
-
- Levy M, Kolodziejczyk AA, Thaiss CA & Elinav E. Dysbiosis and the immune system. Nat. Rev. Immunol 17, 219–232 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UL1 TR001442/TR/NCATS NIH HHS/United States
- P42 ES010337/ES/NIEHS NIH HHS/United States
- RG/18/13/33946/BHF_/British Heart Foundation/United Kingdom
- P30 DK120515/DK/NIDDK NIH HHS/United States
- MR/L003120/1/MRC_/Medical Research Council/United Kingdom
- R01 DK124318/DK/NIDDK NIH HHS/United States
- U01 DK061734/DK/NIDDK NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 DK121378/DK/NIDDK NIH HHS/United States
- CSO_/Chief Scientist Office/United Kingdom
- RG/13/13/30194/BHF_/British Heart Foundation/United Kingdom
- R01 DK106419/DK/NIDDK NIH HHS/United States
