

SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes
- PMID: 35115713
- PMCID: PMC8885846
- DOI: 10.1038/s43018-021-00316-3
SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes
Abstract
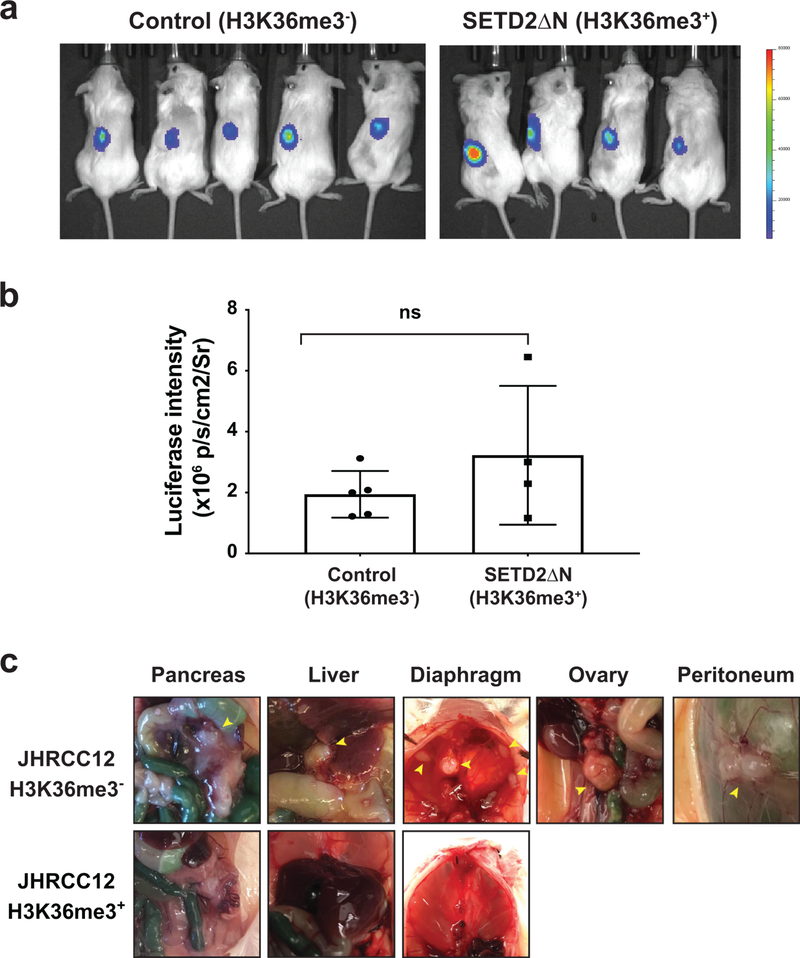
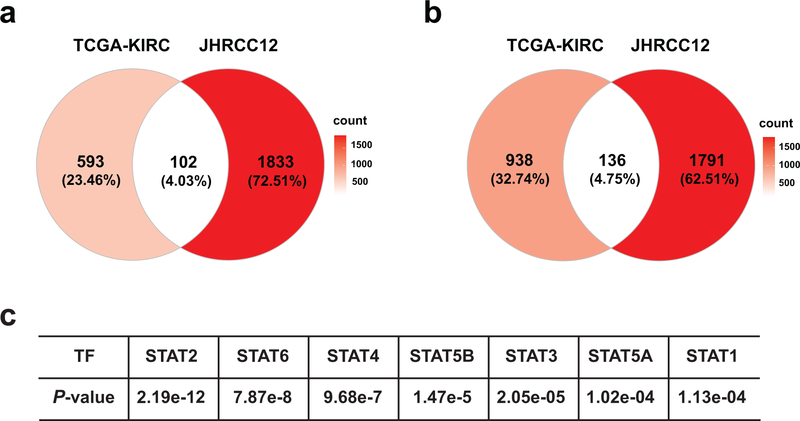
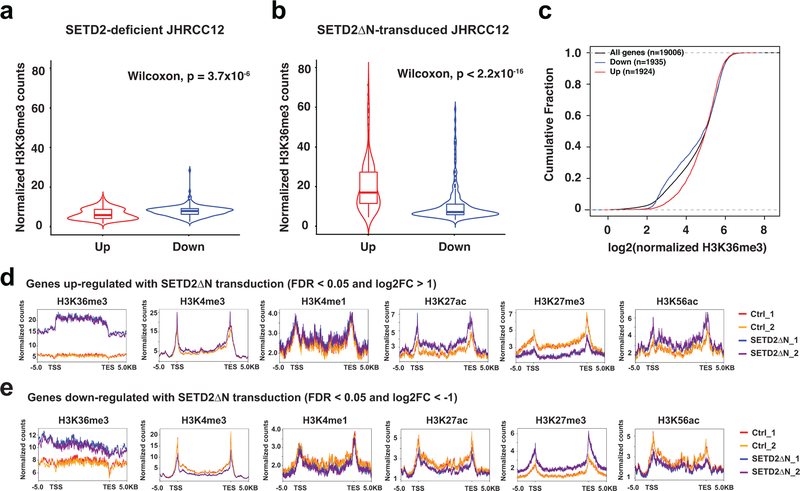
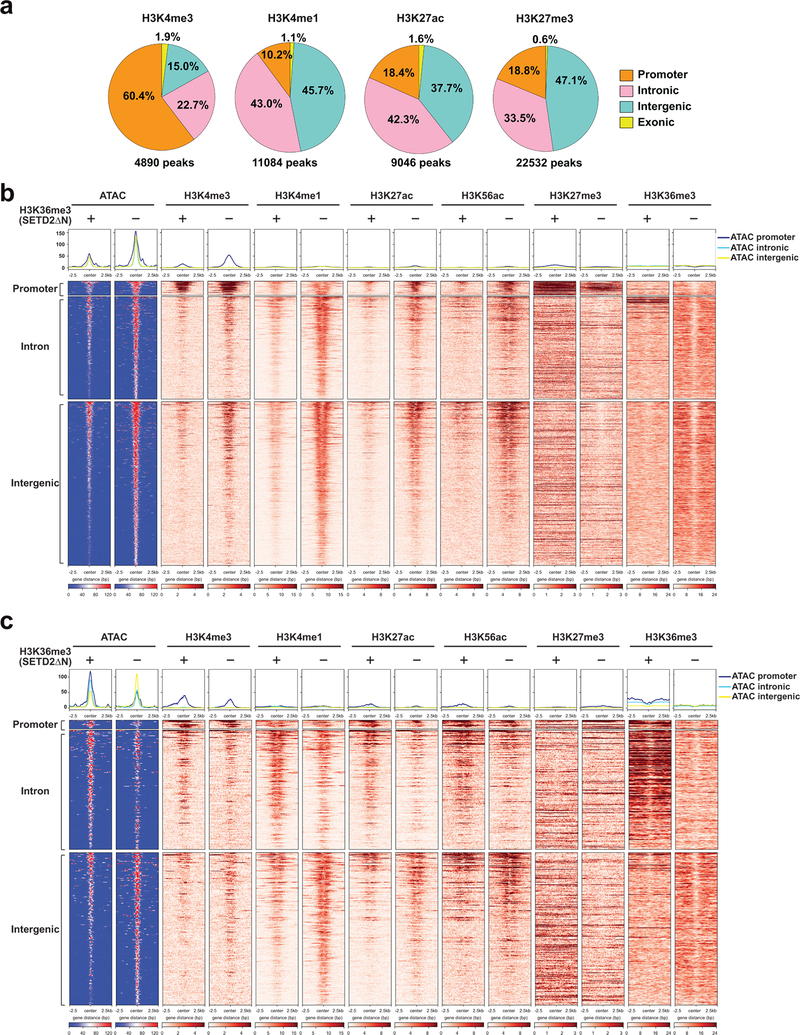
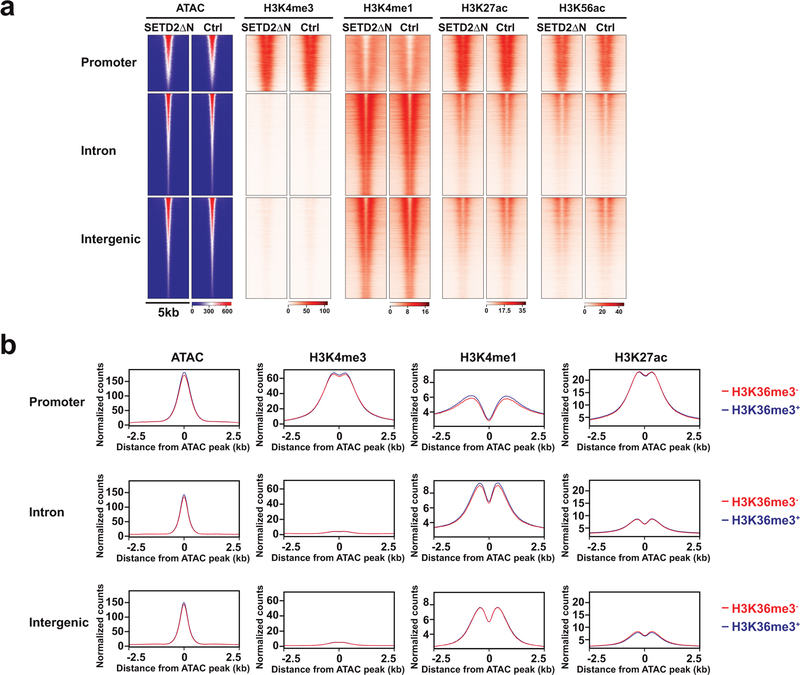
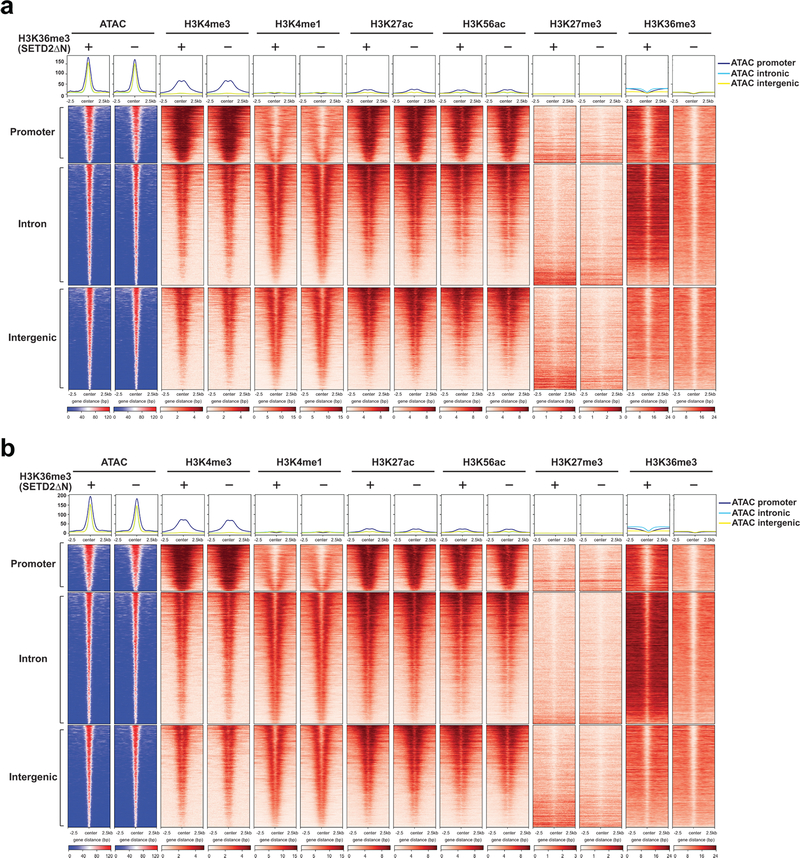
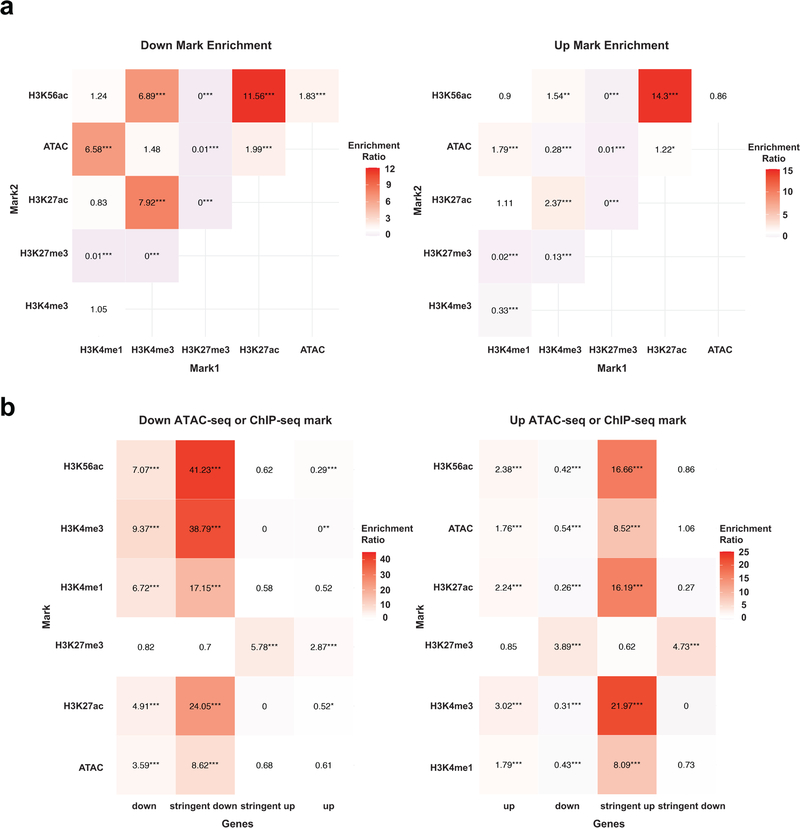
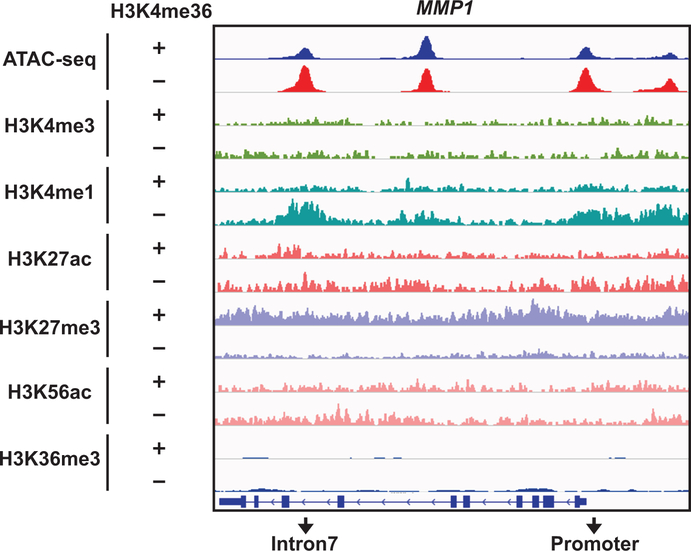
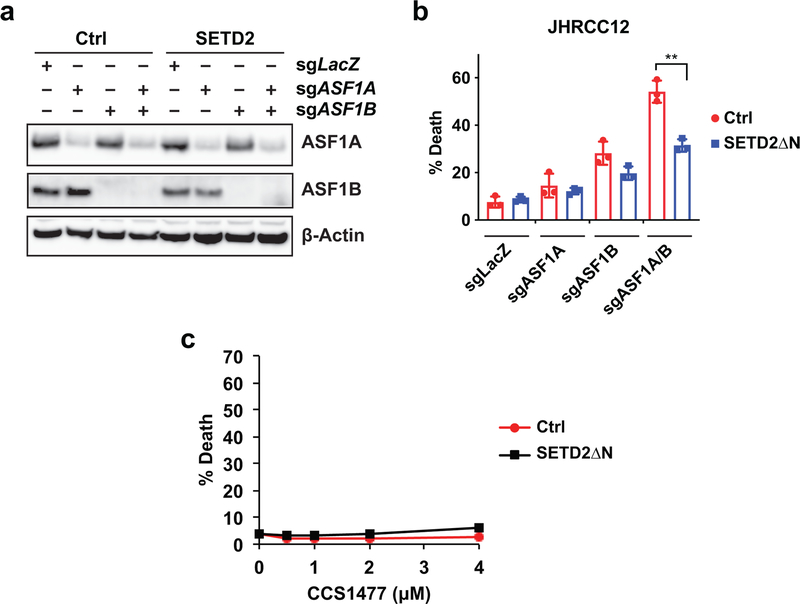
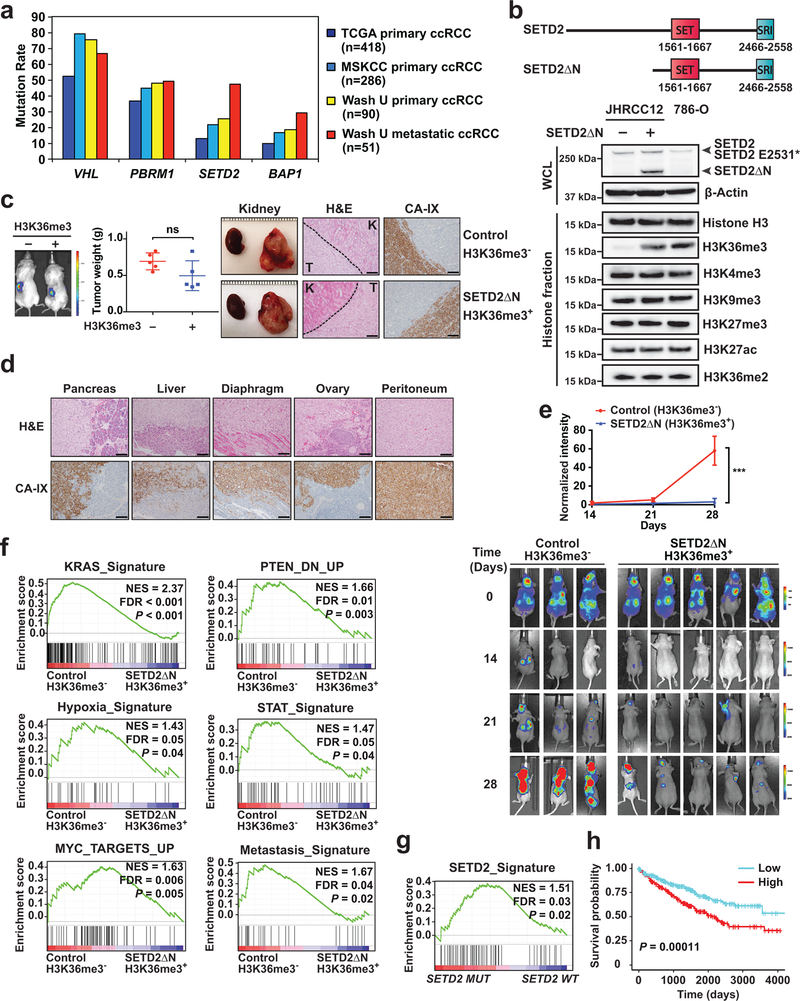
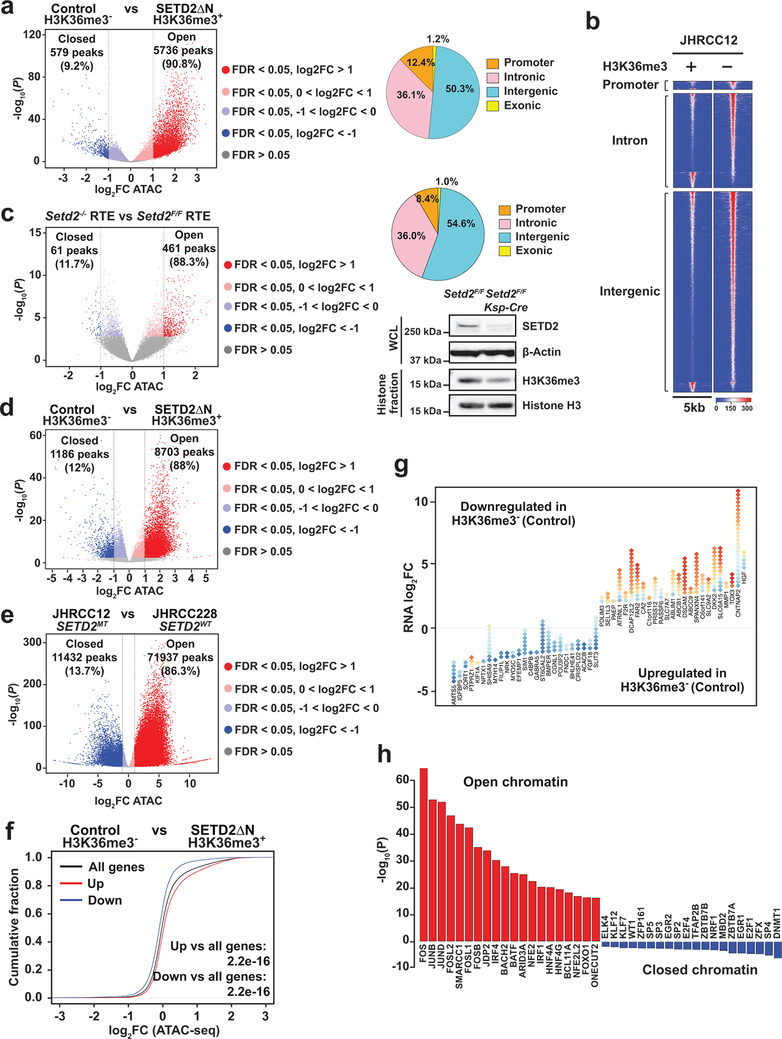
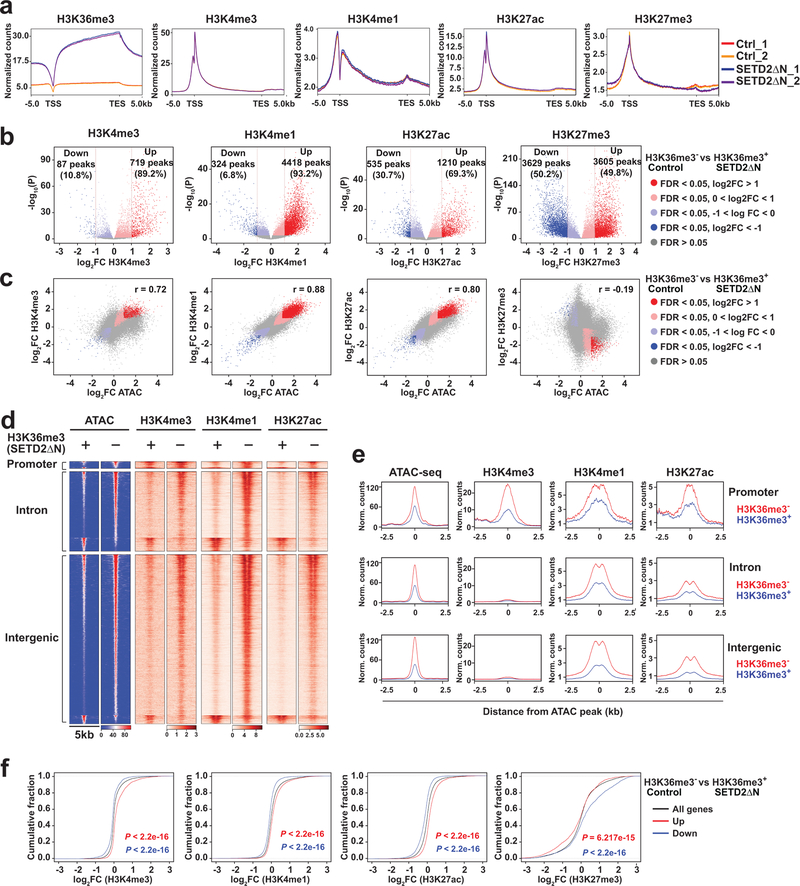
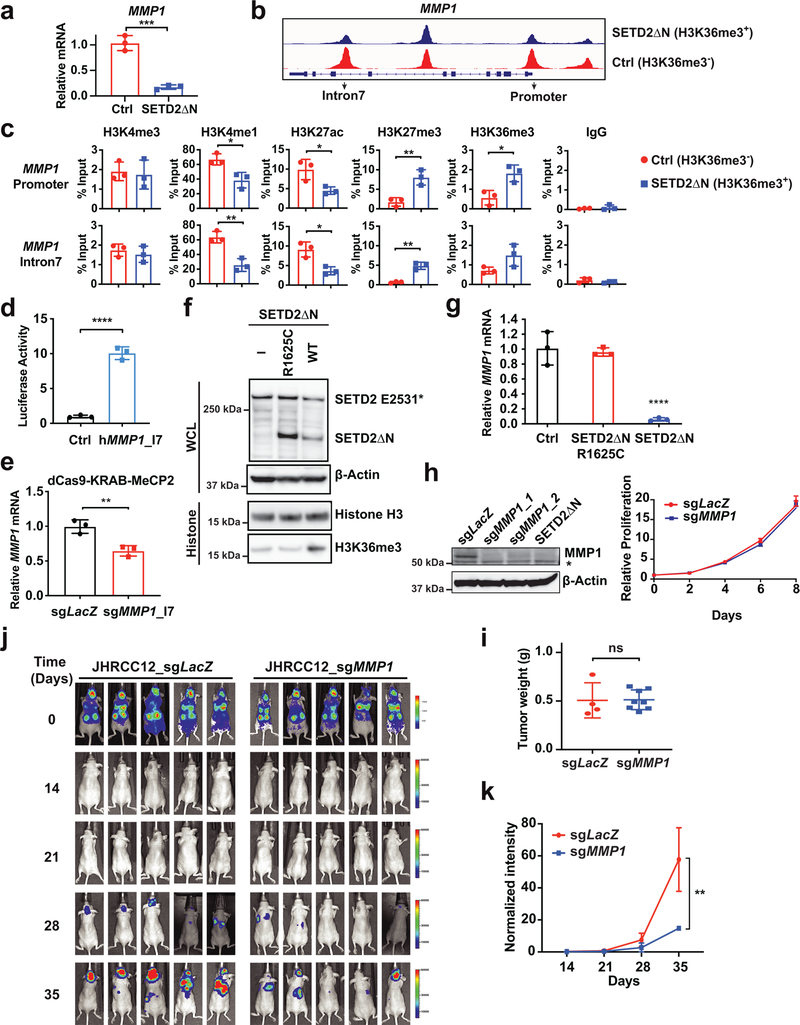
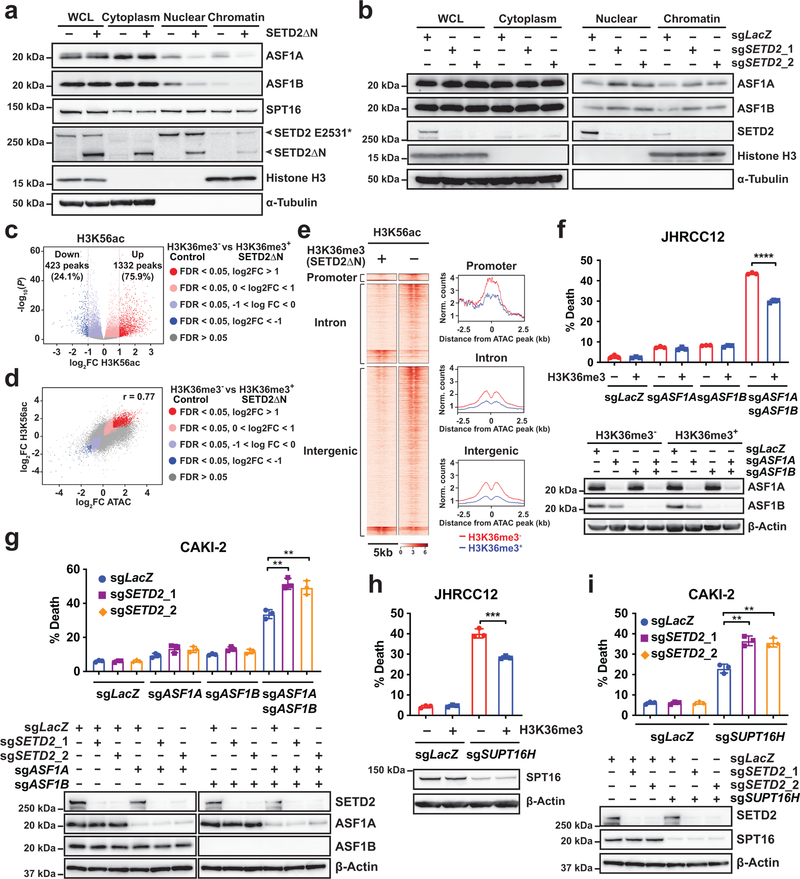
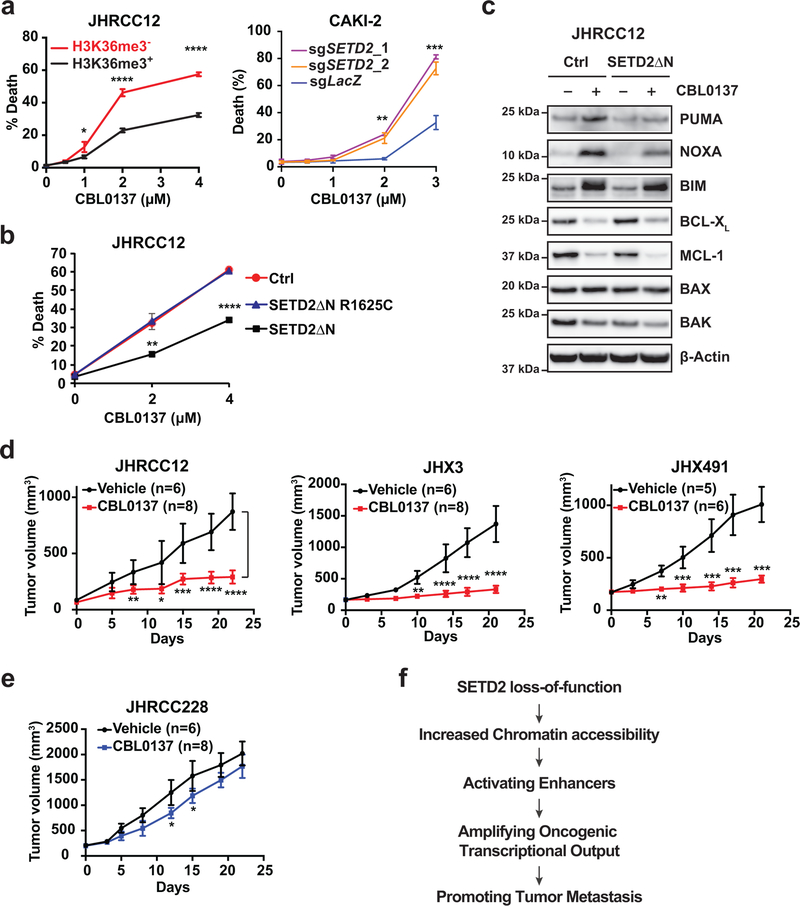
SETD2 is a histone H3 lysine 36 (H3K36) trimethyltransferase that is mutated with high prevalence (13%) in clear cell renal cell carcinoma (ccRCC). Genomic profiling of primary ccRCC tumors reveals a positive correlation between SETD2 mutations and metastasis. However, whether and how SETD2 loss promotes metastasis remains unclear. In this study, we used a SETD2-mutant (SETD2MT) metastatic ccRCC human-derived cell line and xenograft models and showed that H3K36me3 restoration greatly reduced distant metastases of ccRCC in mice in a matrix metalloproteinase 1 (MMP1)-dependent manner. An integrated multiomics analysis using assay for transposase-accessible chromatin using sequencing (ATAC-seq), chromatin immunoprecipitation-sequencing (ChIP-seq) and RNA sequencing (RNA-seq) established a tumor suppressor model in which loss of SETD2-mediated H3K36me3 activates enhancers to drive oncogenic transcriptional output through regulation of chromatin accessibility. Furthermore, we uncovered mechanism-based therapeutic strategies for SETD2-deficient cancer through the targeting of specific histone chaperone complexes, including ASF1A/ASF1B and SPT16. Overall, SETD2 loss creates a permissive epigenetic landscape for cooperating oncogenic drivers to amplify transcriptional output, providing unique therapeutic opportunities.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures

References
-
- Sun X-J et al. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. Journal of Biological Chemistry 280, 35261–35271 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
