

Population-level deficit of homozygosity unveils CPSF3 as an intellectual disability syndrome gene
- PMID: 35121750
- PMCID: PMC8817032
- DOI: 10.1038/s41467-022-28330-8
Population-level deficit of homozygosity unveils CPSF3 as an intellectual disability syndrome gene
Abstract
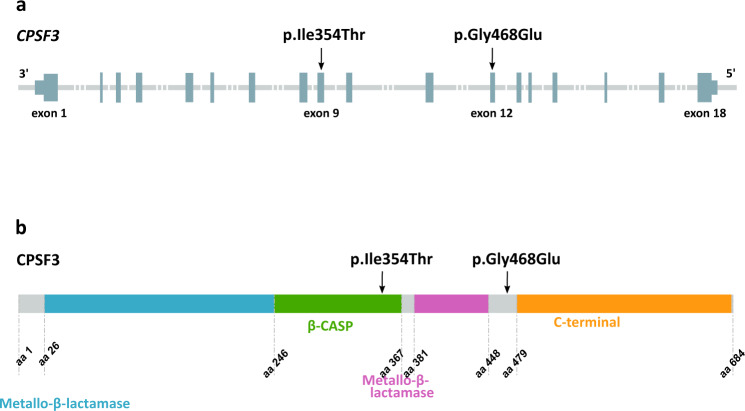
Predicting the pathogenicity of biallelic missense variants can be challenging. Here, we use a deficit of observed homozygous carriers of missense variants, versus an expected number in a set of 153,054 chip-genotyped Icelanders, to identify potentially pathogenic genotypes. We follow three missense variants with a complete deficit of homozygosity and find that their pathogenic effect in homozygous state ranges from severe childhood disease to early embryonic lethality. One of these variants is in CPSF3, a gene not previously linked to disease. From a set of clinically sequenced Icelanders, and by sequencing archival samples targeted through the Icelandic genealogy, we find four homozygous carriers. Additionally, we find two homozygous carriers of Mexican descent of another missense variant in CPSF3. All six homozygous carriers of missense variants in CPSF3 show severe intellectual disability, seizures, microcephaly, and abnormal muscle tone. Here, we show how the absence of certain homozygous genotypes from a large population set can elucidate causes of previously unexplained recessive diseases and early miscarriage.
© 2022. The Author(s).
Conflict of interest statement
Authors affiliated with deCODE genetics/Amgen declare competing interests as employees. The remaining authors declare no competing interests.
Figures
References
-
- Allendorf FW. Genetic drift and the loss of alleles versus heterozygosity. Zoo. Biol. 1986;5:181–190.
-
- Gudbjartsson DF, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015;47:435–444. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
