

Amplifying the spectrum of SPAST gene mutations
- PMID: 35132972
- PMCID: PMC10523053
- DOI: 10.23750/abm.v92iS1.11608
Amplifying the spectrum of SPAST gene mutations
Abstract
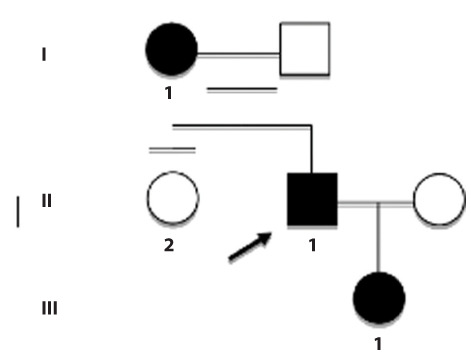
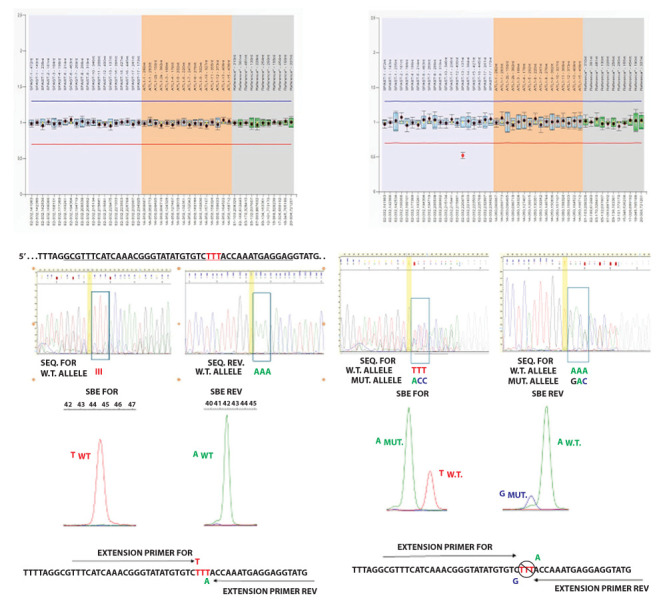
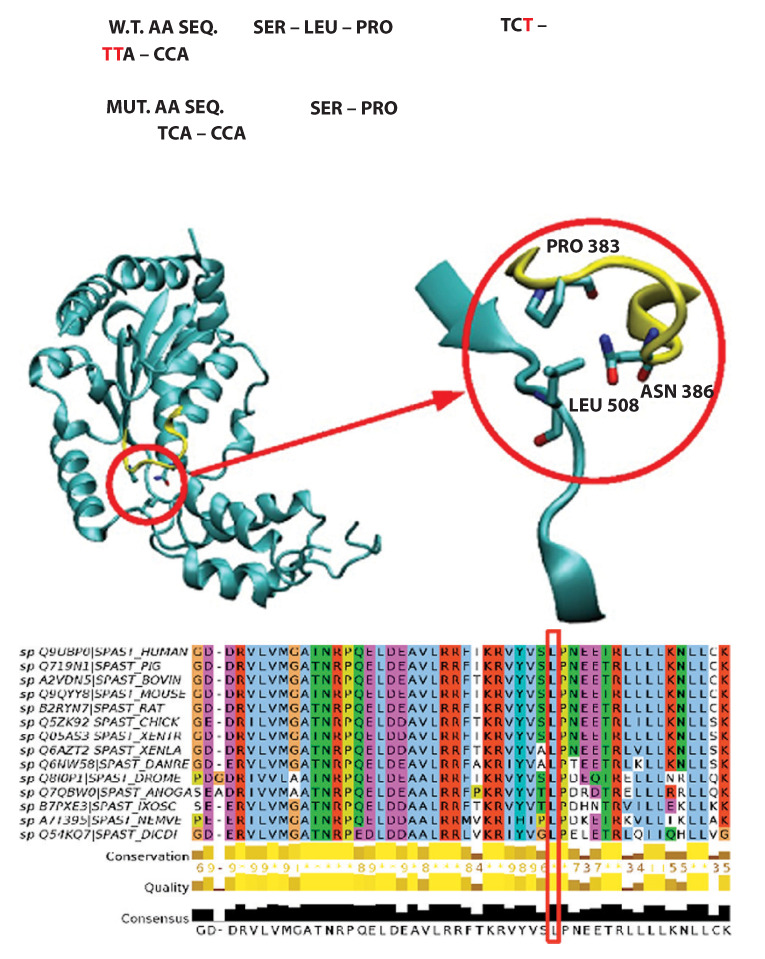
Hereditary spastic paraplegias (HSPs) include a group of neurodegenerative disorders characterized by slowly progressive spasticity and weakness of the lower extremities, caused by axon degeneration of corticospinal tracts. Spastic paraplegia type 4 (SPG4) is the most common autosomal dominant form of HSP and is caused by mutations in the SPAST gene. SPAST gene encodes for the protein spastin, a member of the ATPases Associated with a variety of cellular Activity (AAA) family.We describe a newly variant in SPAST gene, within an Italian family affected by pure HSP. In particular, we found a heterozygous intragenic microdeletion of 3T in exon 13 of SPG4 gene. The 3T deletion results in a mutated protein with a unique leucine residues deletion at the protein position 508, in the AAA ATPase domain. This variant is not registered in any public database either as rare normal variant nor as mutation in SPAST gene and the importance of this aminoacid is confirmed by the absolute conservation in multiple alignments with diverse species. We conclude that the novel SPAST gene variant identified is probably pathogenic and destabilizes the precise arrangement of the nucleotide binding domain, with a consequent loss-of-function of the mutated spastin protein.
Conflict of interest statement
Each author declares that he or she has no commercial associations (e.g. consultancies, stock ownership, equity interest, patent/licensing arrangement etc.) that might pose a conflict of interest in connection with the submitted article.
Figures
Similar articles
-
Four mutations of the spastin gene in Japanese families with spastic paraplegia.J Hum Genet. 2006;51(8):711-715. doi: 10.1007/s10038-006-0412-7. Epub 2006 Jun 21. J Hum Genet. 2006. PMID: 16788734
-
A novel missense mutation in SPAST causes hereditary spastic paraplegia in male members of a family: A case report.Mol Med Rep. 2023 Apr;27(4):79. doi: 10.3892/mmr.2023.12966. Epub 2023 Feb 24. Mol Med Rep. 2023. PMID: 36825575 Free PMC article.
-
Hereditary spastic paraplegia SPG4: what is known and not known about the disease.Brain. 2015 Sep;138(Pt 9):2471-84. doi: 10.1093/brain/awv178. Epub 2015 Jun 20. Brain. 2015. PMID: 26094131 Free PMC article. Review.
-
Novel SPAST Deletion Mutation in an American Family With Hereditary Spastic Paraplegia: A Case Report.J Investig Med High Impact Case Rep. 2025 Jan-Dec;13:23247096251323173. doi: 10.1177/23247096251323173. J Investig Med High Impact Case Rep. 2025. PMID: 40019011 Free PMC article.
-
Exon 8-17 deletions of SPAST in a Chinese family with hereditary spastic paraplegia: a case report and literature review.J Neurol Sci. 2015 Oct 15;357(1-2):282-4. doi: 10.1016/j.jns.2015.07.003. Epub 2015 Jul 3. J Neurol Sci. 2015. PMID: 26165777 Review.
Cited by
-
A novel variant (p.A524P) in Spastin is responsible for a Chinese family with hereditary spastic paraplegia.Mol Biol Rep. 2024 Sep 4;51(1):951. doi: 10.1007/s11033-024-09880-0. Mol Biol Rep. 2024. PMID: 39230614
-
Large-scale whole-exome sequencing of neuropsychiatric diseases and traits in 350,770 adults.Nat Hum Behav. 2024 Jun;8(6):1194-1208. doi: 10.1038/s41562-024-01861-4. Epub 2024 Apr 8. Nat Hum Behav. 2024. PMID: 38589703
-
ATXN2 polyglutamine intermediate repeats length expansions in Malaysian patients with amyotrophic lateral sclerosis (ALS).Neurogenetics. 2025 Jan 13;26(1):19. doi: 10.1007/s10048-024-00798-0. Neurogenetics. 2025. PMID: 39804470
References
-
- Erfanian Omidvar M, Torkamandi S, Rezaei S, et al. Genotype-phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J Neurol. 2019;268(6):2065–2082. - PubMed
-
- Hazan J, Fonknechten N, Mavel D, et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet. 1999;23(3):296–303. - PubMed
-
- Lumb JH, Connell JW, Allison R, Reid E. The AAA ATPase spastin links microtubule severing to membrane modelling. Biochim Biophys Acta. 2012;1823(1):192–7. - PubMed
-
- Parodi L, Fenu S, Barbier M, et al. SPATAX network. Spastic paraplegia due to SPAST mutations is modified by the underlying mutation and sex. Brain. 2018;141(12):3331–3342. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
