

A single-cell Arabidopsis root atlas reveals developmental trajectories in wild-type and cell identity mutants
- PMID: 35134336
- PMCID: PMC9014886
- DOI: 10.1016/j.devcel.2022.01.008
A single-cell Arabidopsis root atlas reveals developmental trajectories in wild-type and cell identity mutants
Abstract
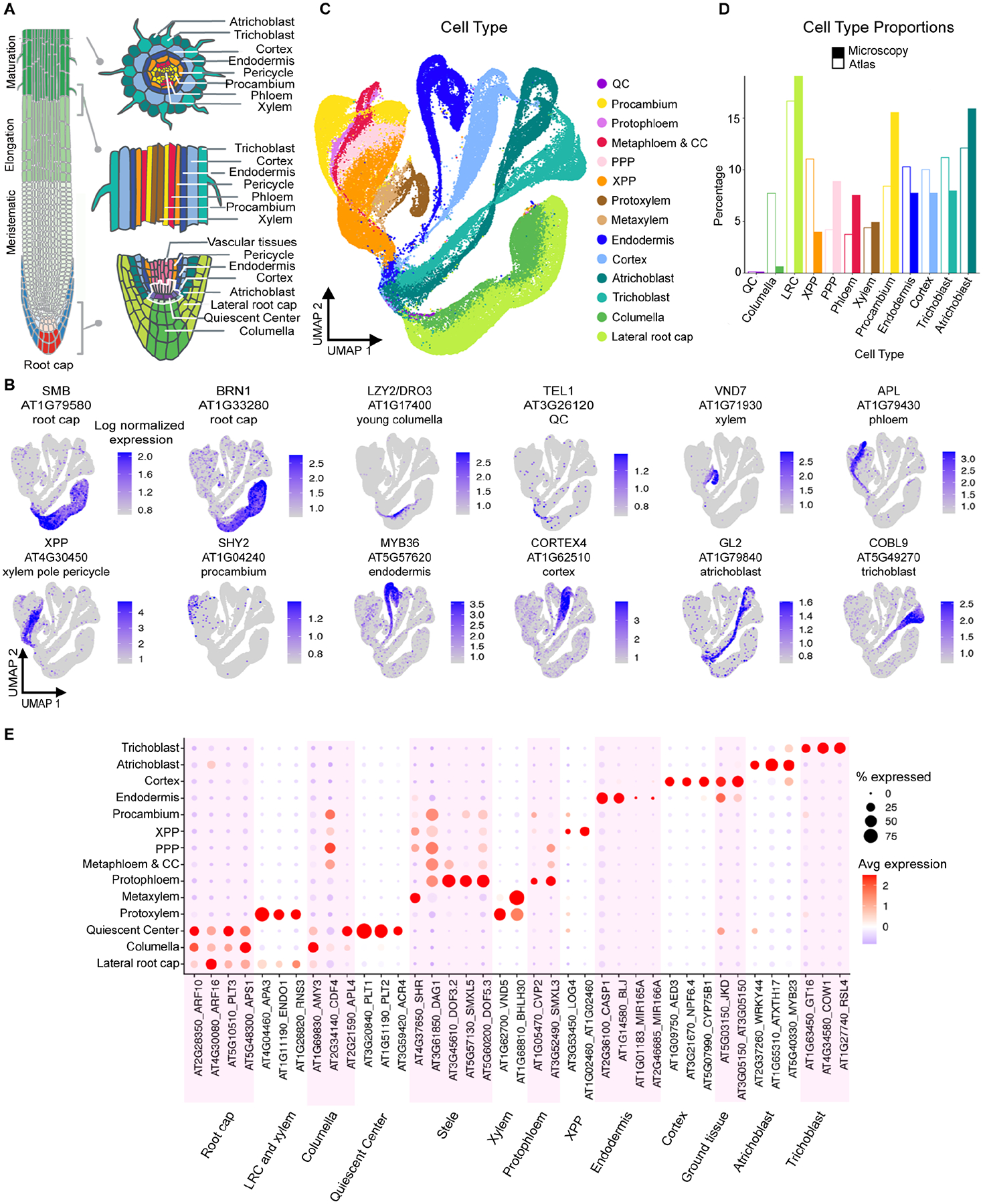
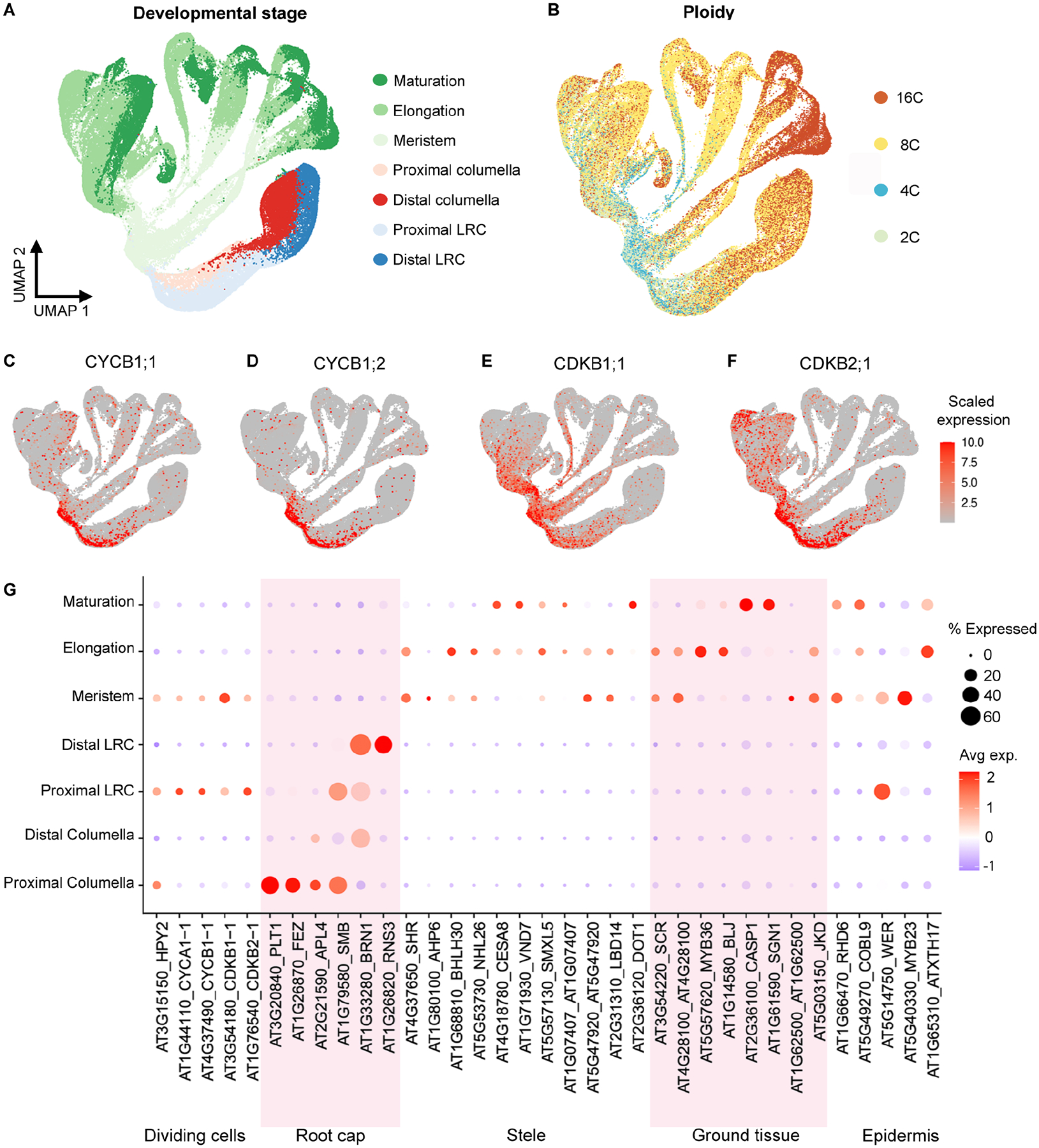
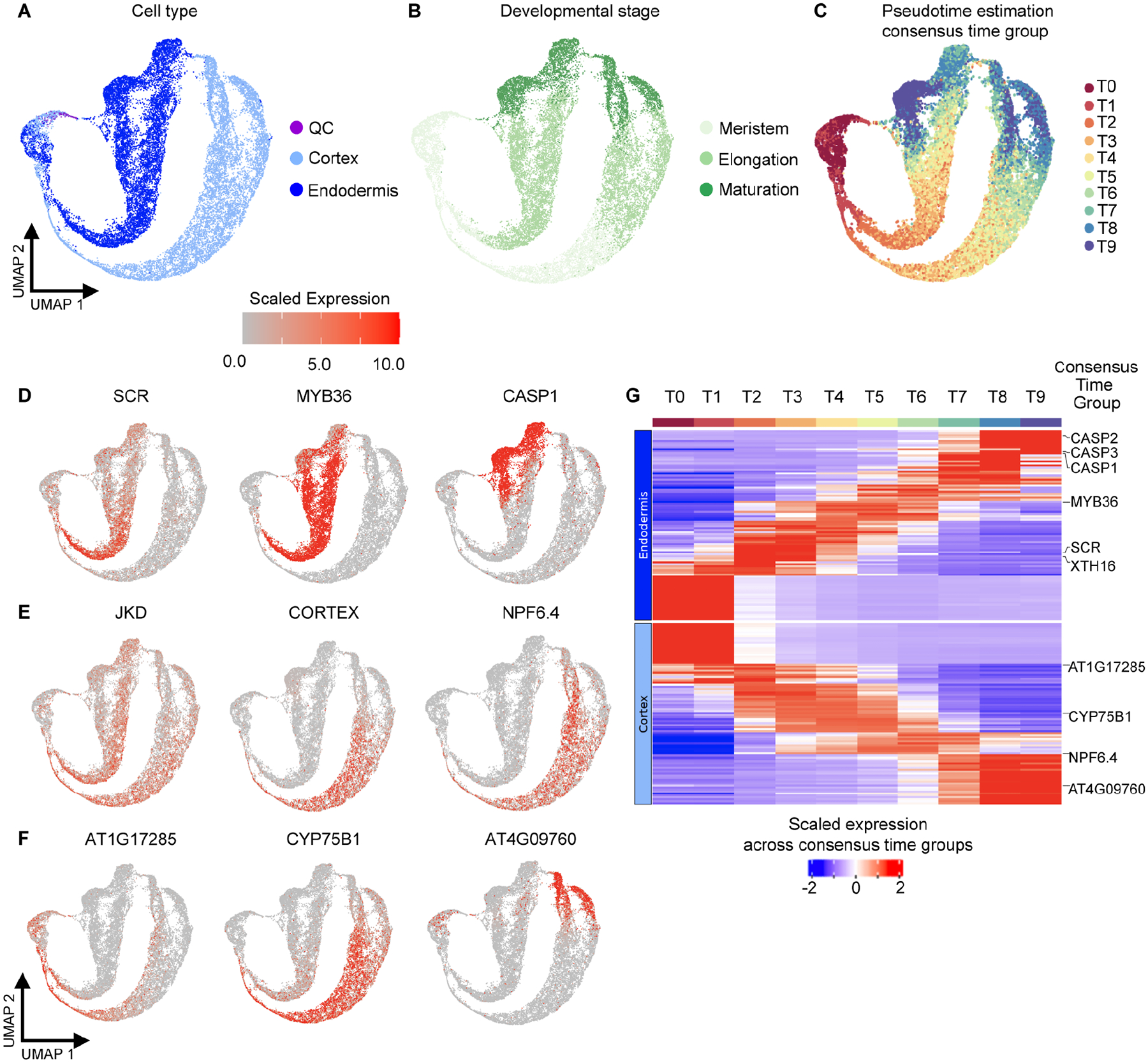
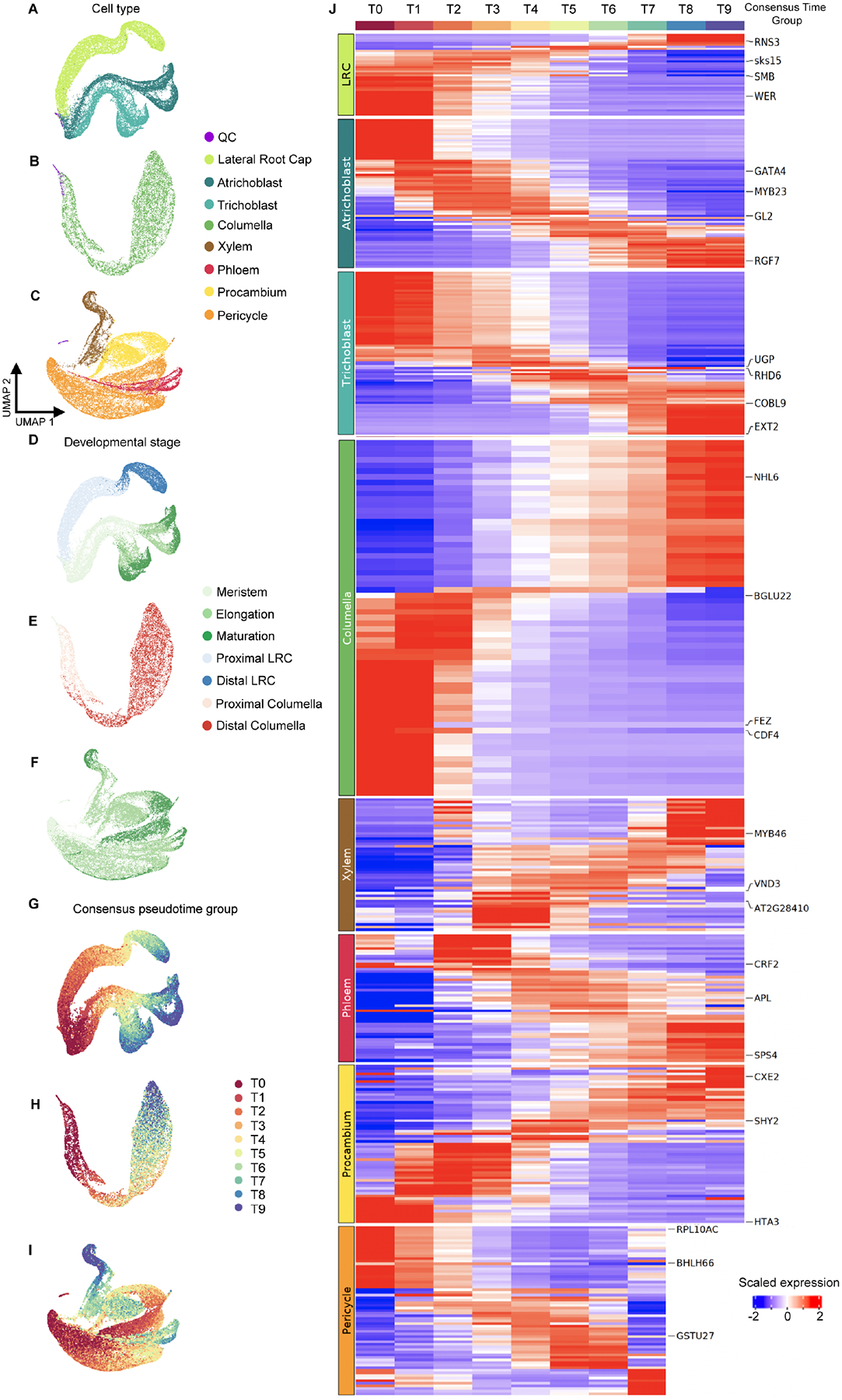
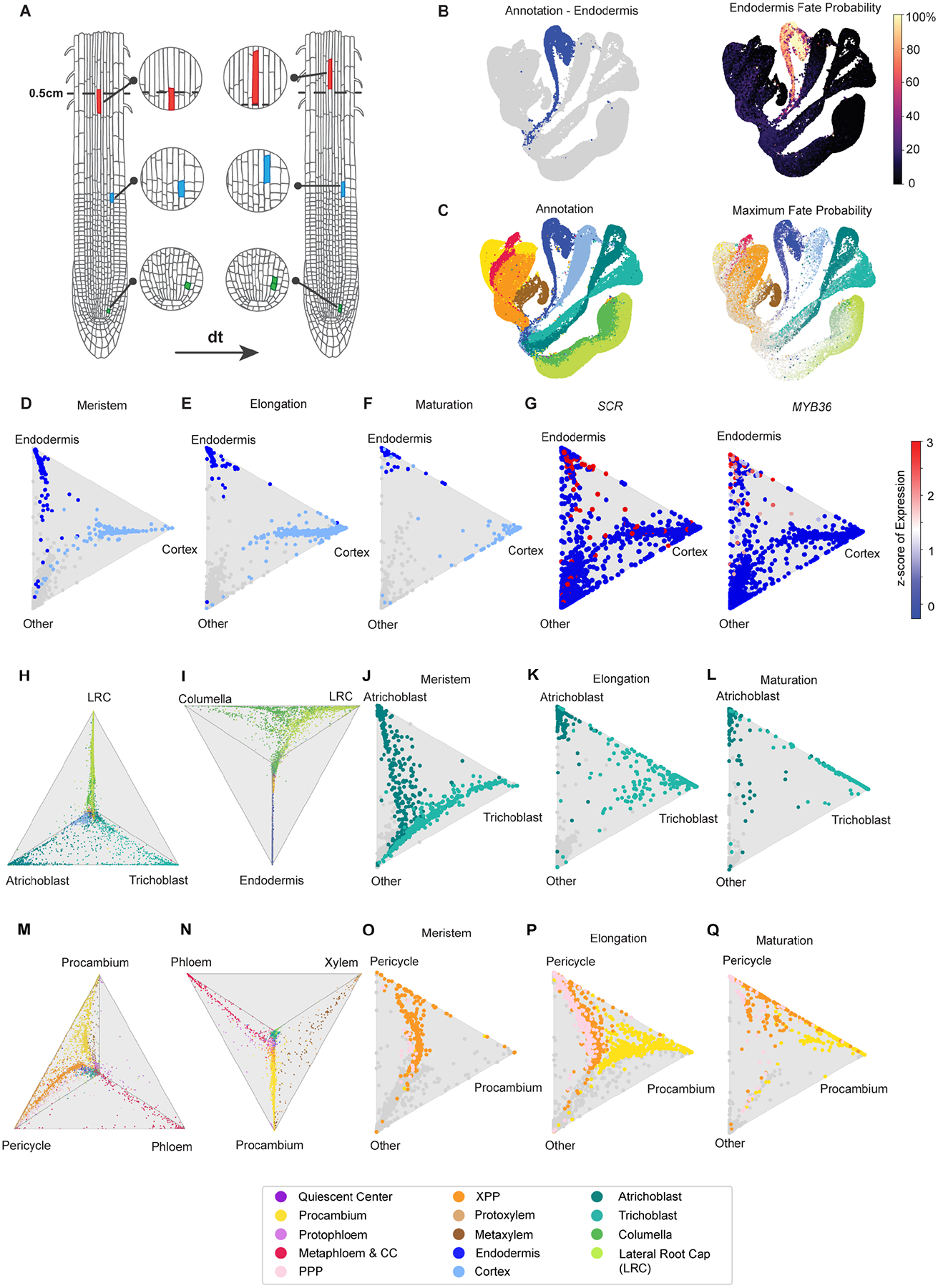
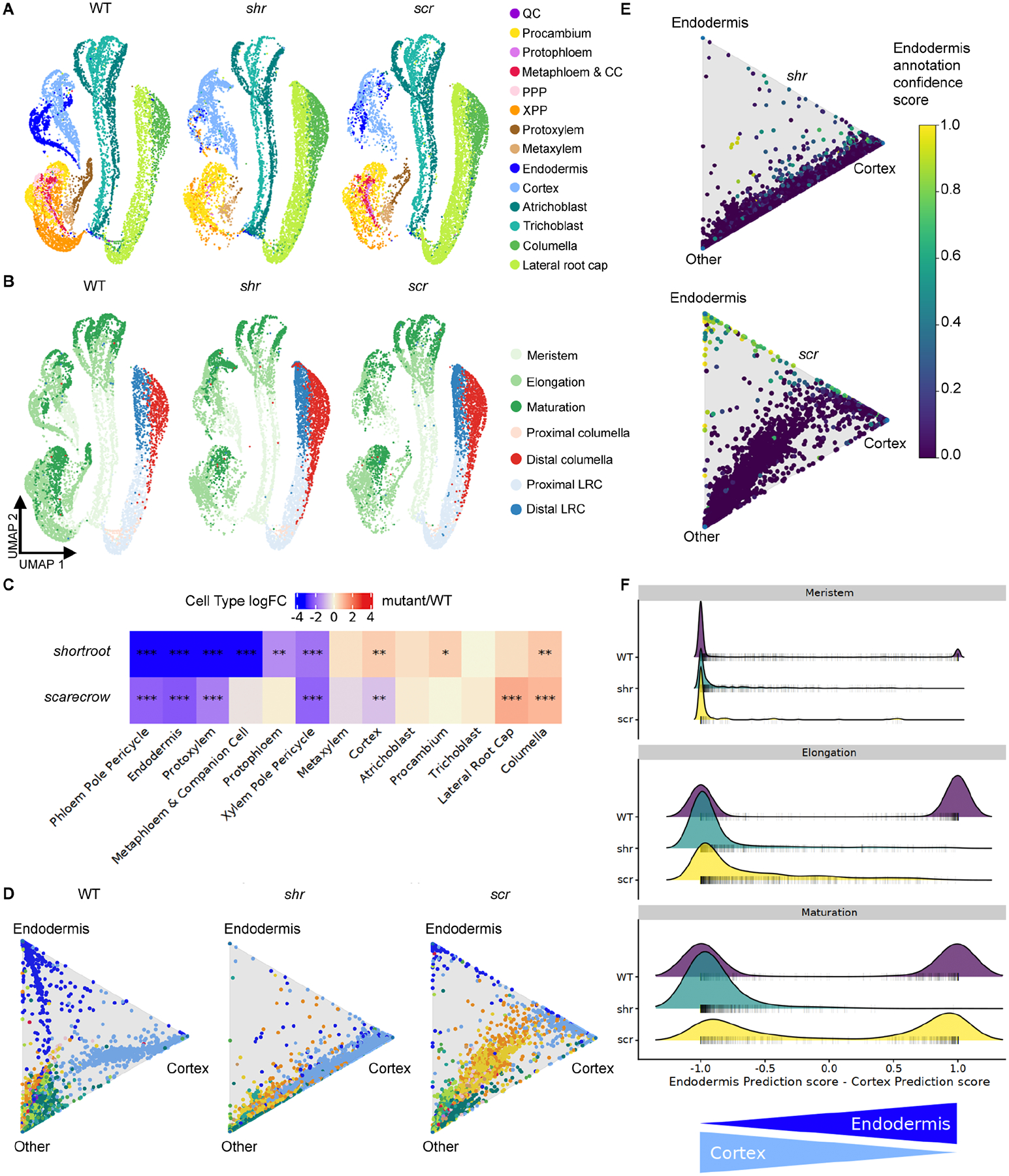
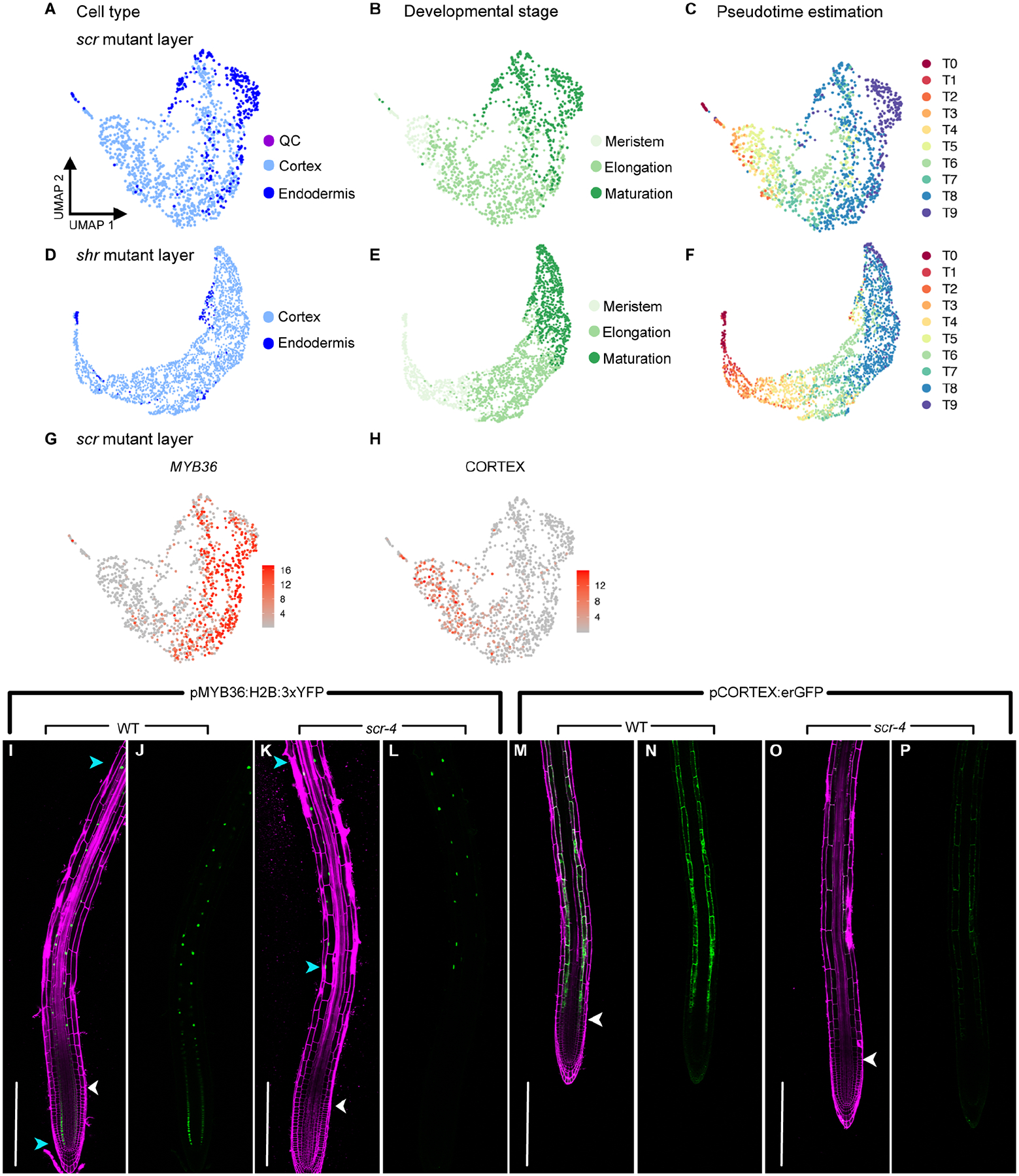
In all multicellular organisms, transcriptional networks orchestrate organ development. The Arabidopsis root, with its simple structure and indeterminate growth, is an ideal model for investigating the spatiotemporal transcriptional signatures underlying developmental trajectories. To map gene expression dynamics across root cell types and developmental time, we built a comprehensive, organ-scale atlas at single-cell resolution. In addition to estimating developmental progressions in pseudotime, we employed the mathematical concept of optimal transport to infer developmental trajectories and identify their underlying regulators. To demonstrate the utility of the atlas to interpret new datasets, we profiled mutants for two key transcriptional regulators at single-cell resolution, shortroot and scarecrow. We report transcriptomic and in vivo evidence for tissue trans-differentiation underlying a mixed cell identity phenotype in scarecrow. Our results support the atlas as a rich community resource for unraveling the transcriptional programs that specify and maintain cell identity to regulate spatiotemporal organ development.
Keywords: Arabidopsis; SCARECROW; SHORTROOT; cell fate; development; root; scRNA-seq; transcriptomics.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests P.N.B. is a member of the Developmental Cell advisory board and is the co-founder and Chair of the Scientific Advisory Board of Hi Fidelity Genetics, a company that works on crop root growth.
Figures
References
-
- Andrews S (2010). FastQC: A quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
