

Analysis of SARS-CoV-2 synonymous codon usage evolution throughout the COVID-19 pandemic
- PMID: 35134624
- PMCID: PMC8808327
- DOI: 10.1016/j.virol.2022.01.011
Analysis of SARS-CoV-2 synonymous codon usage evolution throughout the COVID-19 pandemic
Abstract
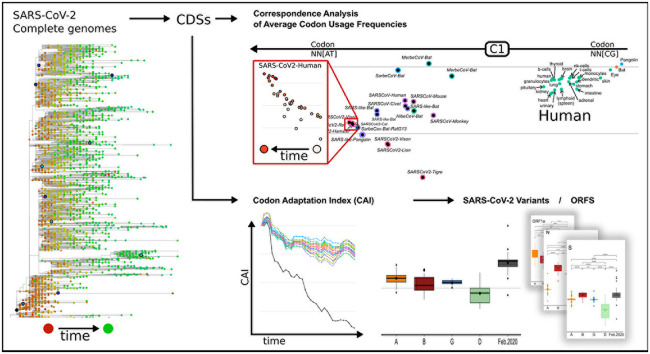
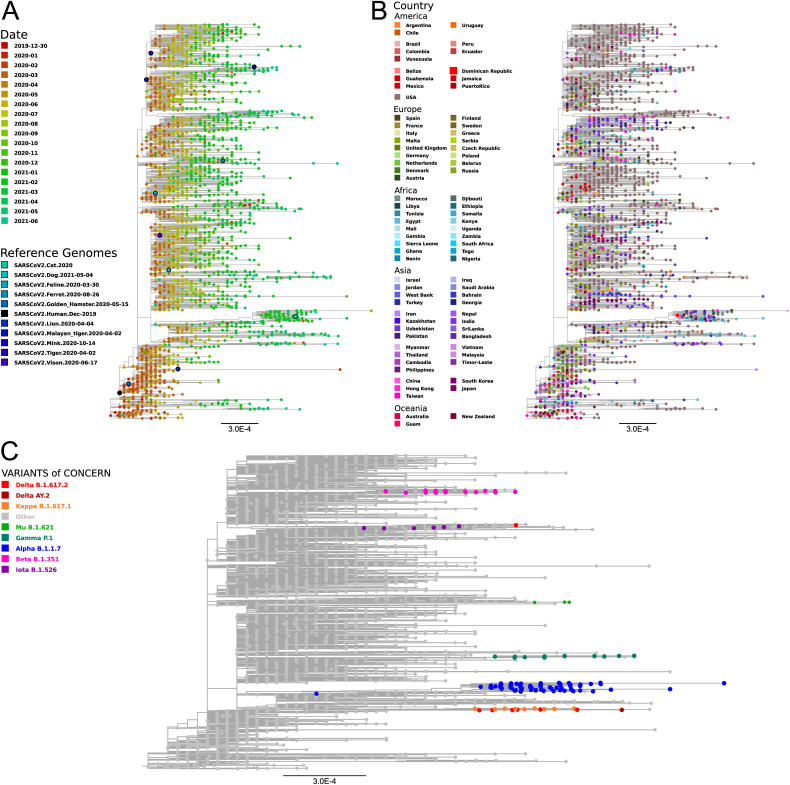
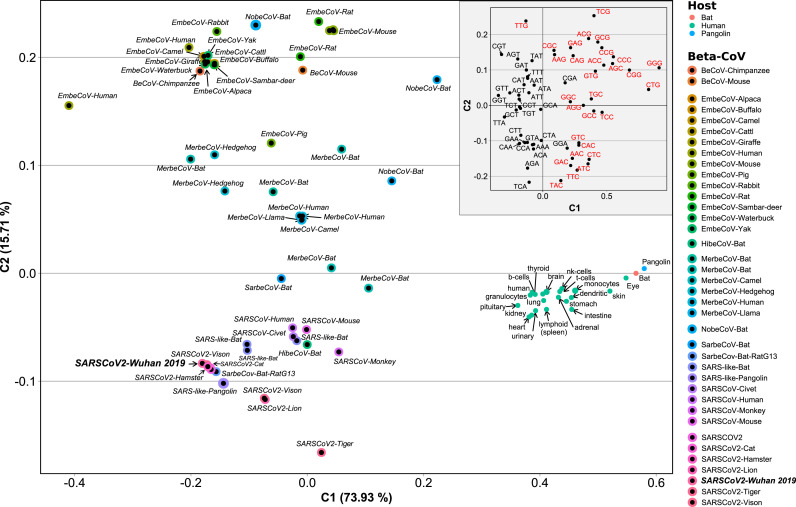
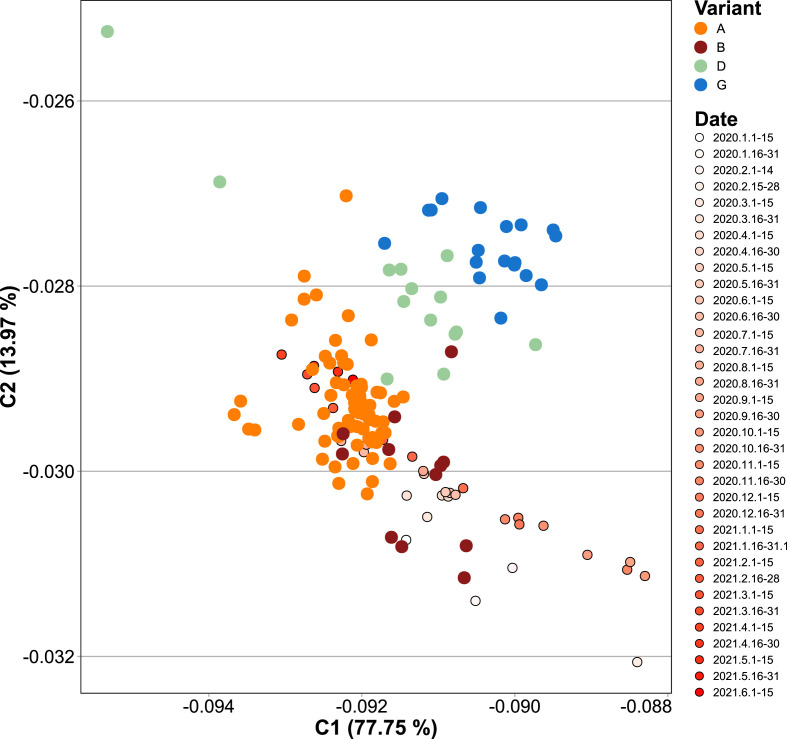
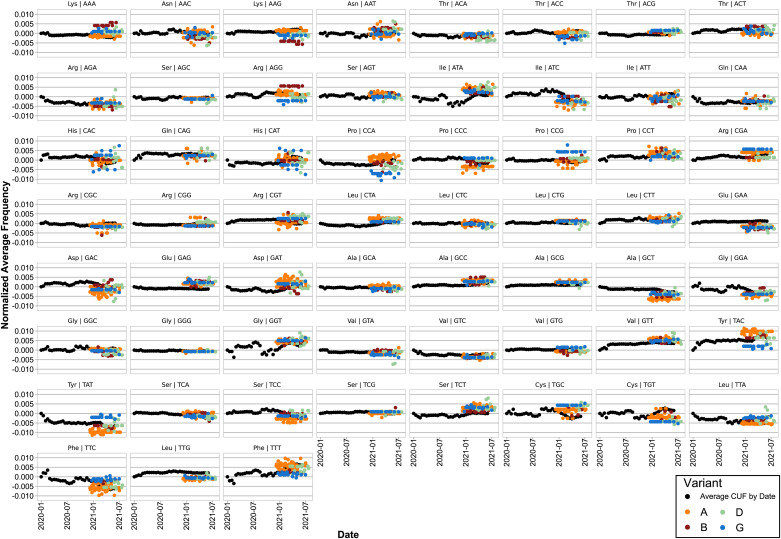
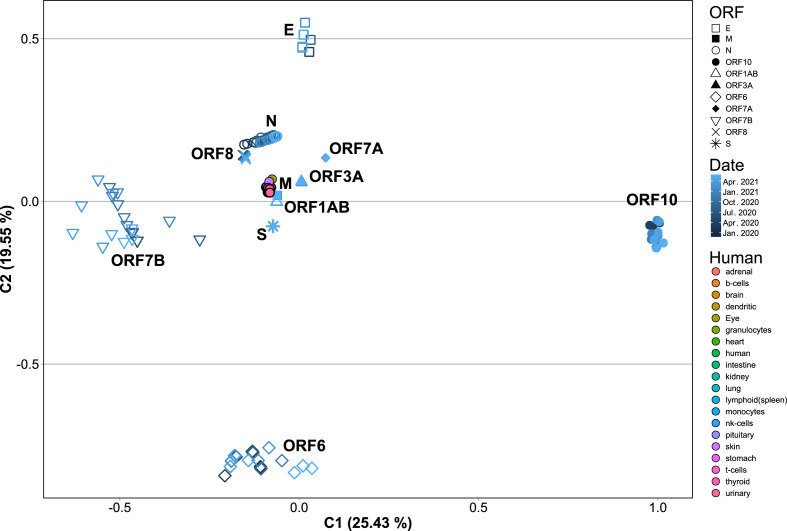
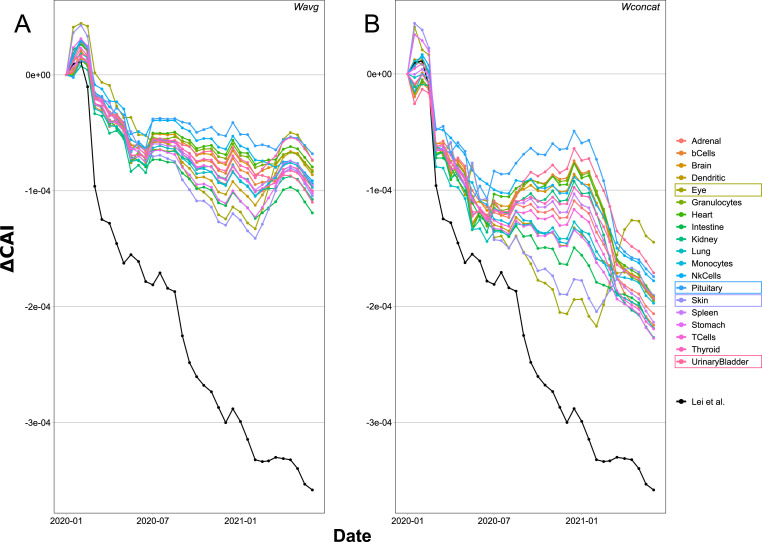
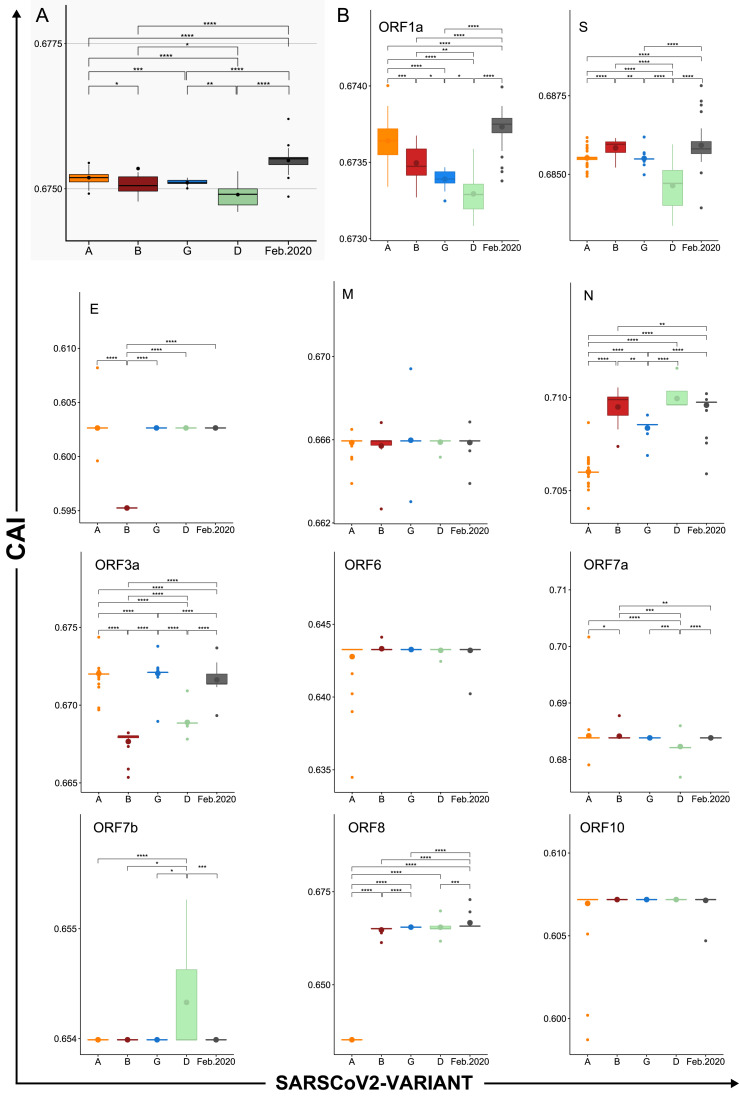
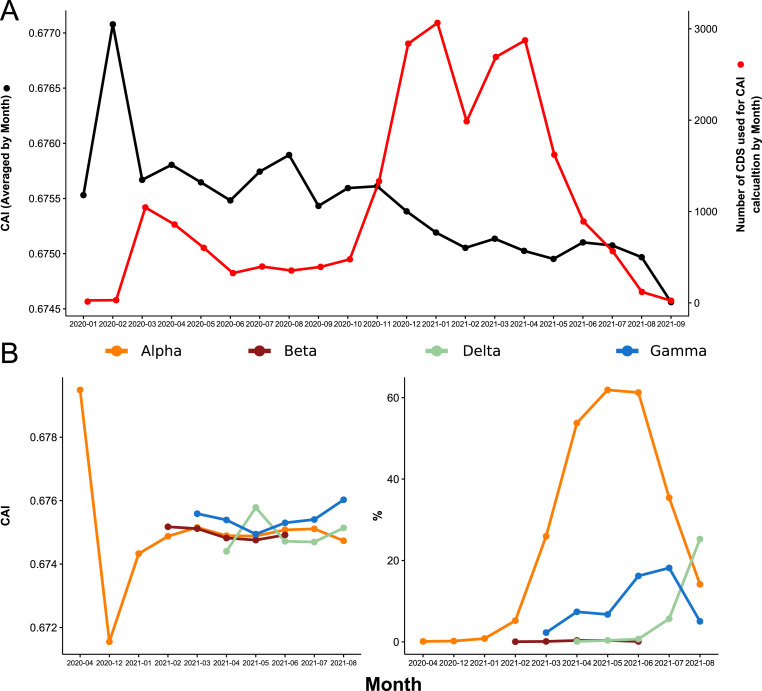
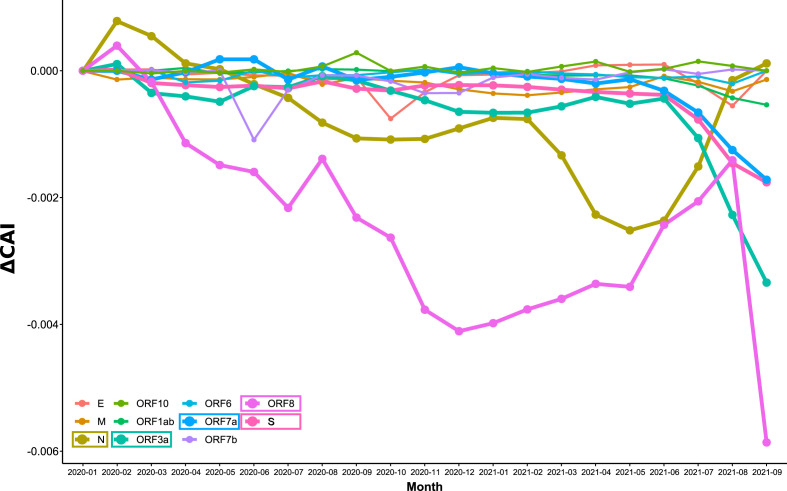
SARS-CoV-2, the seventh coronavirus known to infect humans, can cause severe life-threatening respiratory pathologies. To better understand SARS-CoV-2 evolution, genome-wide analyses have been made, including the general characterization of its codons usage profile. Here we present a bioinformatic analysis of the evolution of SARS-CoV-2 codon usage over time using complete genomes collected since December 2019. Our results show that SARS-CoV-2 codon usage pattern is antagonistic to, and it is getting farther away from that of the human host. Further, a selection of deoptimized codons over time, which was accompanied by a decrease in both the codon adaptation index and the effective number of codons, was observed. All together, these findings suggest that SARS-CoV-2 could be evolving, at least from the perspective of the synonymous codon usage, to become less pathogenic.
Keywords: Betacoronavirus; COVID-19; Codon usage bias; SARS-CoV-2.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
