

Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination
- PMID: 35137080
- PMCID: PMC8882382
- DOI: 10.1093/gbe/evac018
Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination
Abstract
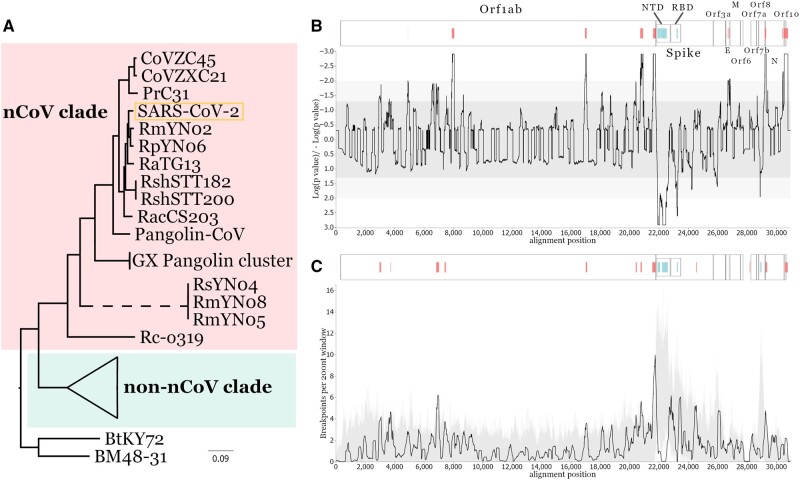
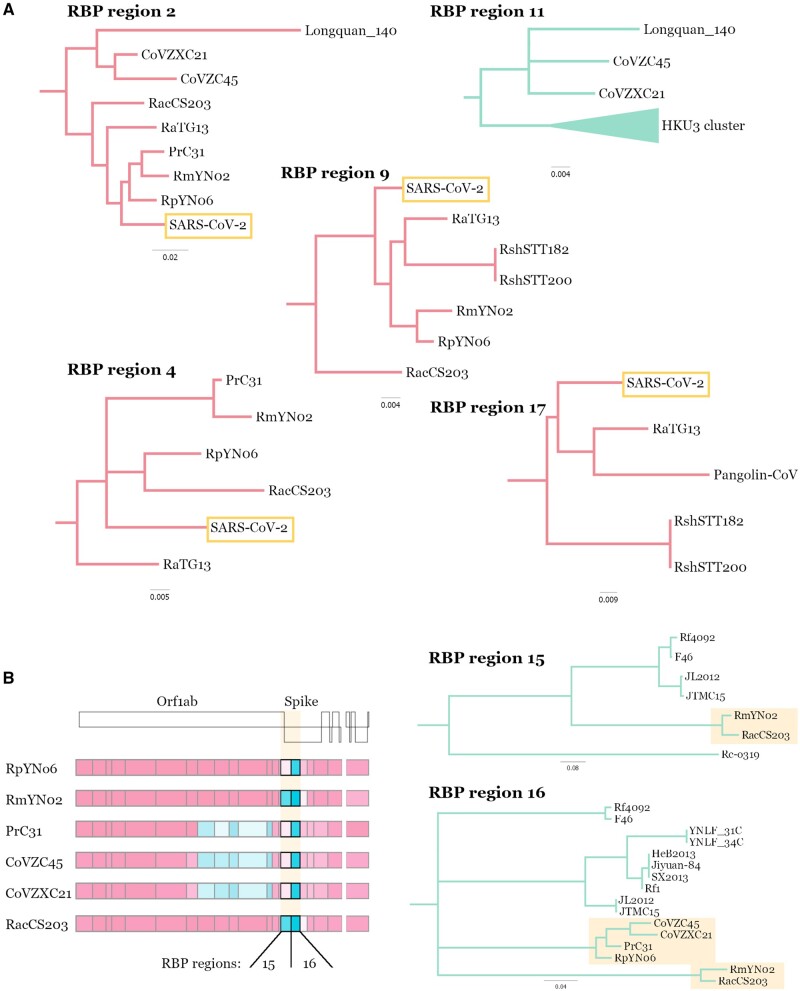
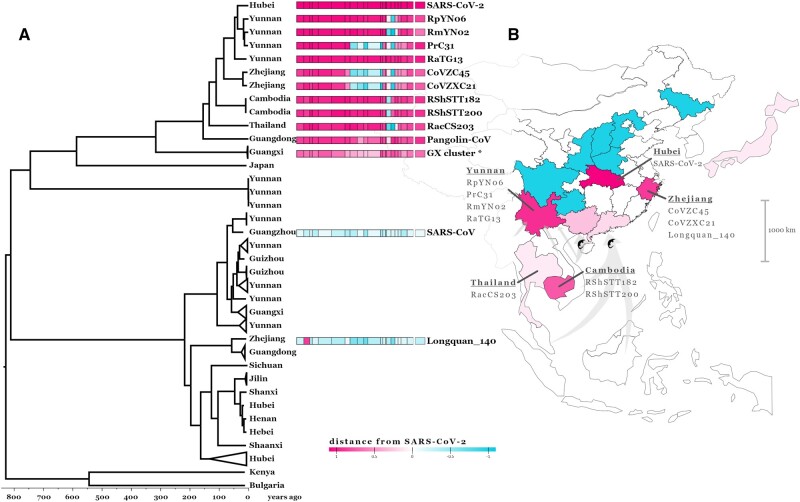
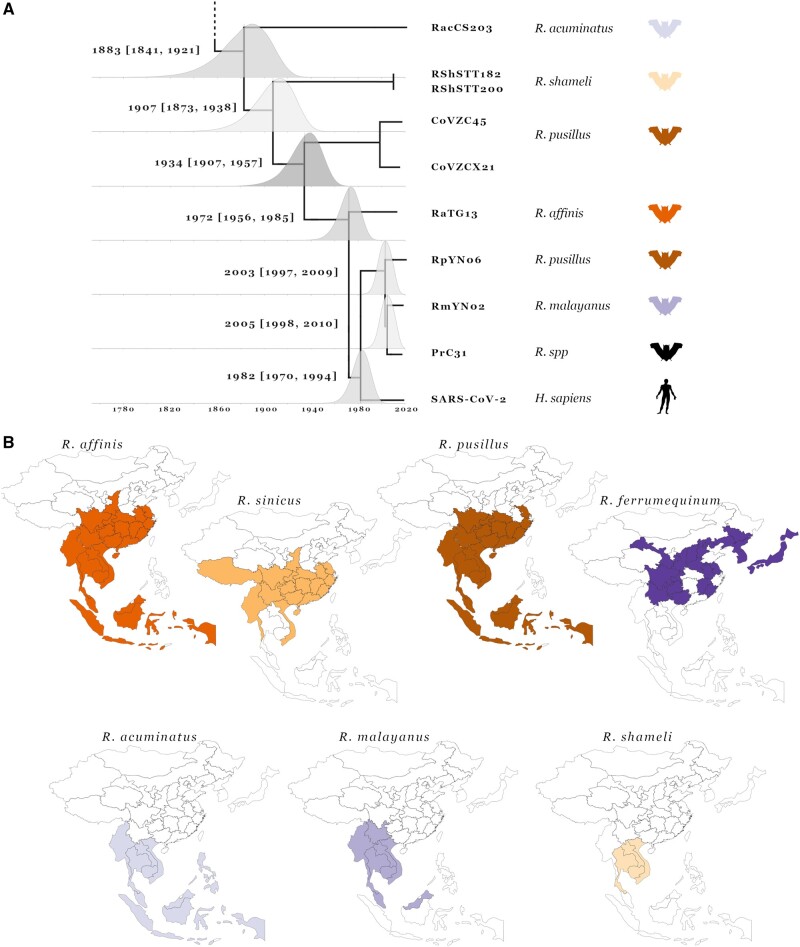
The lack of an identifiable intermediate host species for the proximal animal ancestor of SARS-CoV-2, and the large geographical distance between Wuhan and where the closest evolutionary related coronaviruses circulating in horseshoe bats (members of the Sarbecovirus subgenus) have been identified, is fueling speculation on the natural origins of SARS-CoV-2. We performed a comprehensive phylogenetic study on SARS-CoV-2 and all the related bat and pangolin sarbecoviruses sampled so far. Determining the likely recombination events reveals a highly reticulate evolutionary history within this group of coronaviruses. Distribution of the inferred recombination events is nonrandom with evidence that Spike, the main target for humoral immunity, is beside a recombination hotspot likely driving antigenic shift events in the ancestry of bat sarbecoviruses. Coupled with the geographic ranges of their hosts and the sampling locations, across southern China, and into Southeast Asia, we confirm that horseshoe bats, Rhinolophus, are the likely reservoir species for the SARS-CoV-2 progenitor. By tracing the recombinant sequence patterns, we conclude that there has been relatively recent geographic movement and cocirculation of these viruses' ancestors, extending across their bat host ranges in China and Southeast Asia over the last 100 years. We confirm that a direct proximal ancestor to SARS-CoV-2 has not yet been sampled, since the closest known relatives collected in Yunnan shared a common ancestor with SARS-CoV-2 approximately 40 years ago. Our analysis highlights the need for dramatically more wildlife sampling to: 1) pinpoint the exact origins of SARS-CoV-2's animal progenitor, 2) the intermediate species that facilitated transmission from bats to humans (if there is one), and 3) survey the extent of the diversity in the related sarbecoviruses' phylogeny that present high risk for future spillovers.
Keywords: Rhinolophus; Sarbecoviruses; COVID-19; SARS-CoV-2; bats; coronaviruses; host range; origin; pangolins; recombination.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Akaike H. 1998. Information theory and an extension of the maximum likelihood principle. In: Parzen E, Tanabe K, Kitagawa G, editors. Selected Papers of Hirotugu Akaike. Springer Series in Statistics (Perspectives in Statistics). New York: Springer. p. 199–213.
-
- Bates P, Bumrungsri S, Csorba G, Soisook P.. 2019. Rhinolophus malayanus. IUCN Red List Threat. Species 2019. [Internet]:e.T19551A21978424. International Union for Conservation of Nature and Natural Resources (IUCN). Available from: https://www.iucnredlist.org/species/19551/21978424.
-
- Boni MF, et al. 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 5(11):1408–1417. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
