

Flap Dynamics in Pepsin-Like Aspartic Proteases: A Computational Perspective Using Plasmepsin-II and BACE-1 as Model Systems
- PMID: 35138093
- PMCID: PMC8889585
- DOI: 10.1021/acs.jcim.1c00840
Flap Dynamics in Pepsin-Like Aspartic Proteases: A Computational Perspective Using Plasmepsin-II and BACE-1 as Model Systems
Abstract
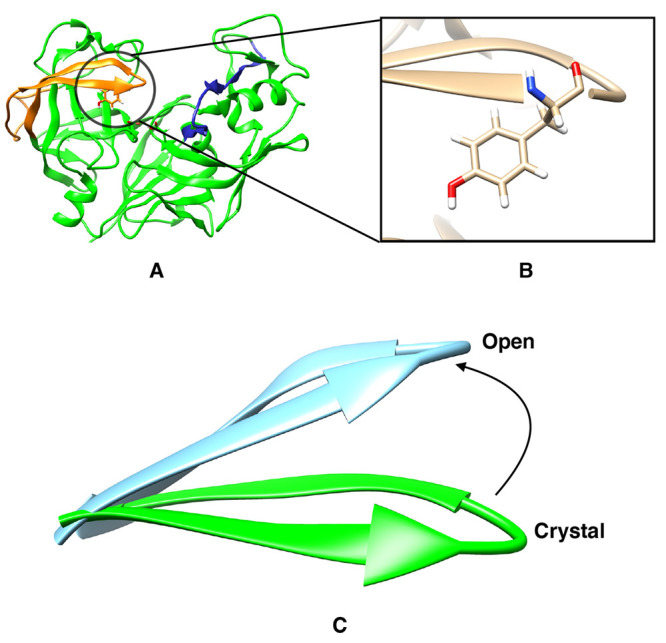
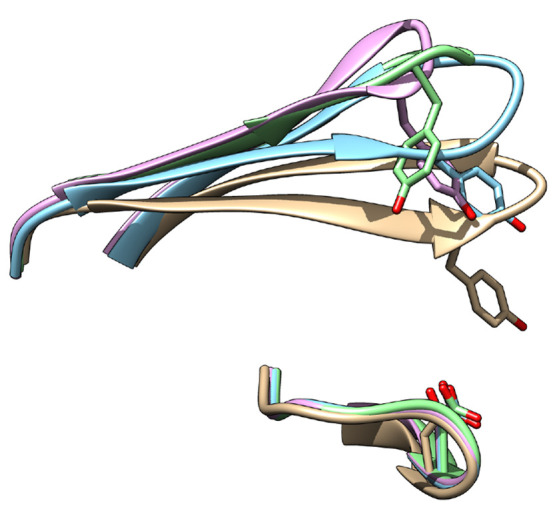
The flexibility of β hairpin structure known as the flap plays a key role in catalytic activity and substrate intake in pepsin-like aspartic proteases. Most of these enzymes share structural and sequential similarity. In this study, we have used apo Plm-II and BACE-1 as model systems. In the apo form of the proteases, a conserved tyrosine residue in the flap region remains in a dynamic equilibrium between the normal and flipped states through rotation of the χ1 and χ2 angles. Independent MD simulations of Plm-II and BACE-1 remained stuck either in the normal or flipped state. Metadynamics simulations using side-chain torsion angles (χ1 and χ2 of tyrosine) as collective variables sampled the transition between the normal and flipped states. Qualitatively, the two states were predicted to be equally populated. The normal and flipped states were stabilized by H-bond interactions to a tryptophan residue and to the catalytic aspartate, respectively. Further, mutation of tyrosine to an amino-acid with smaller side-chain, such as alanine, reduced the flexibility of the flap and resulted in a flap collapse (flap loses flexibility and remains stuck in a particular state). This is in accordance with previous experimental studies, which showed that mutation to alanine resulted in loss of activity in pepsin-like aspartic proteases. Our results suggest that the ring flipping associated with the tyrosine side-chain is the key order parameter that governs flap dynamics and opening of the binding pocket in most pepsin-like aspartic proteases.
Conflict of interest statement
The authors declare no competing financial interest.
Figures





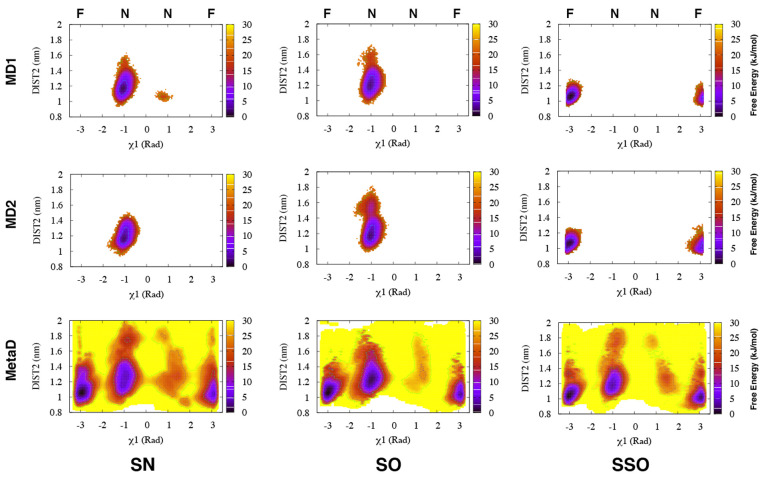
 rad, whereas
simulations starting from
the SSO structure only sampled the flipped (F) state.
Torsion-Metad simulations starting from the SN, SO, or SSO structures
sampled both the N and F states.
rad, whereas
simulations starting from
the SSO structure only sampled the flipped (F) state.
Torsion-Metad simulations starting from the SN, SO, or SSO structures
sampled both the N and F states.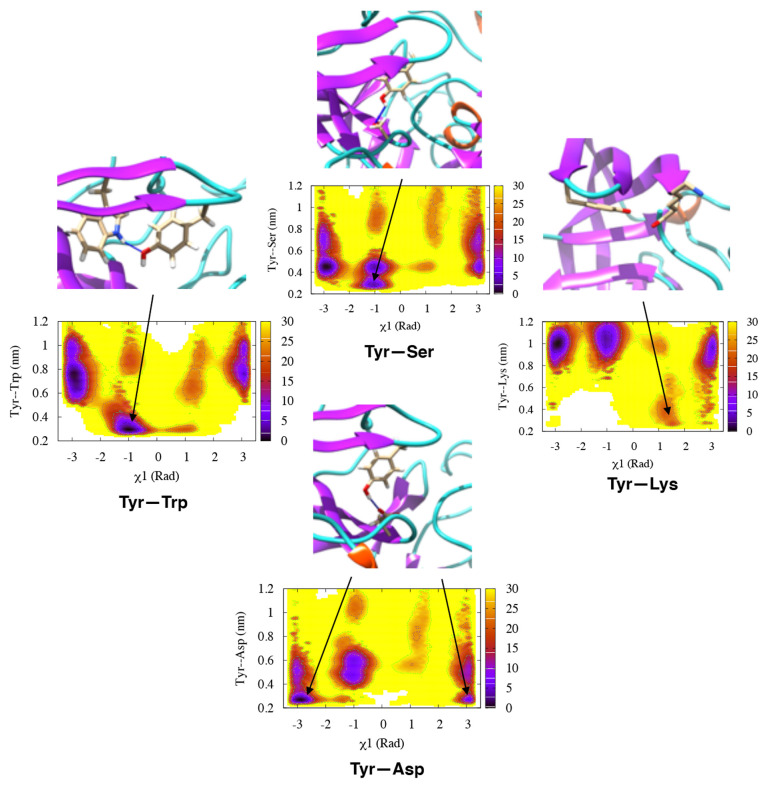




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
