

Screening of ETO2-GLIS2-induced Super Enhancers identifies targetable cooperative dependencies in acute megakaryoblastic leukemia
- PMID: 35138899
- PMCID: PMC8827662
- DOI: 10.1126/sciadv.abg9455
Screening of ETO2-GLIS2-induced Super Enhancers identifies targetable cooperative dependencies in acute megakaryoblastic leukemia
Abstract
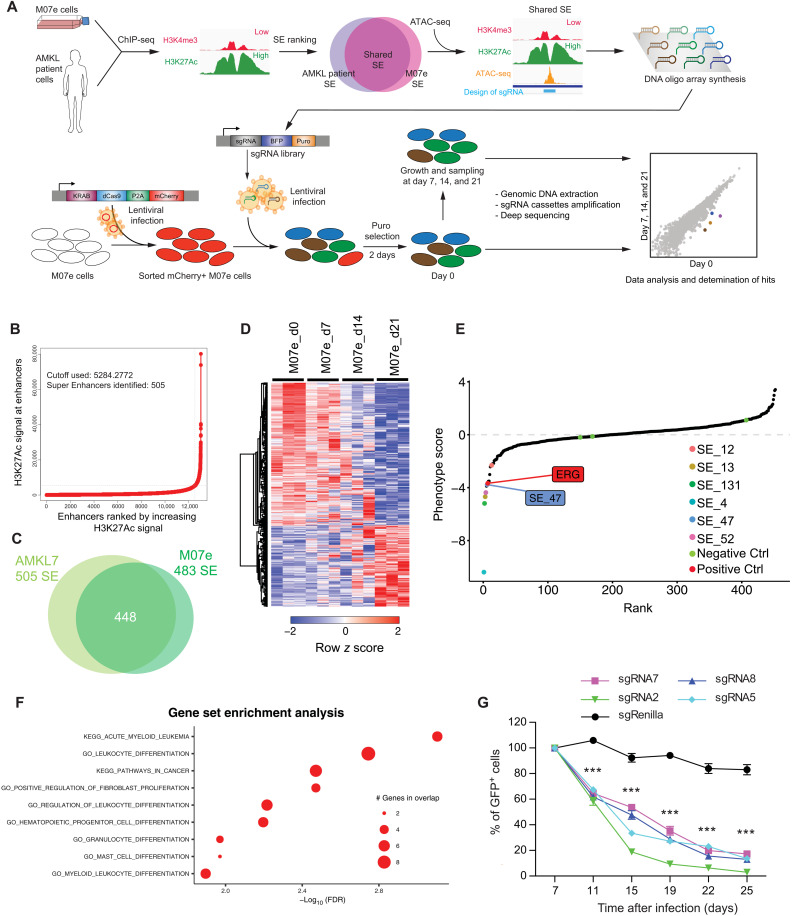
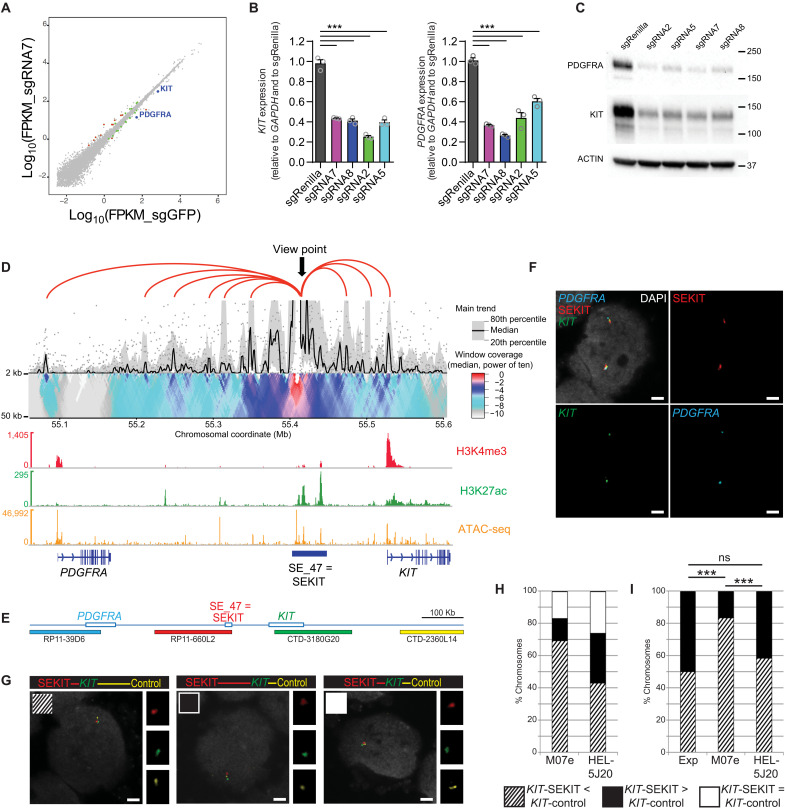
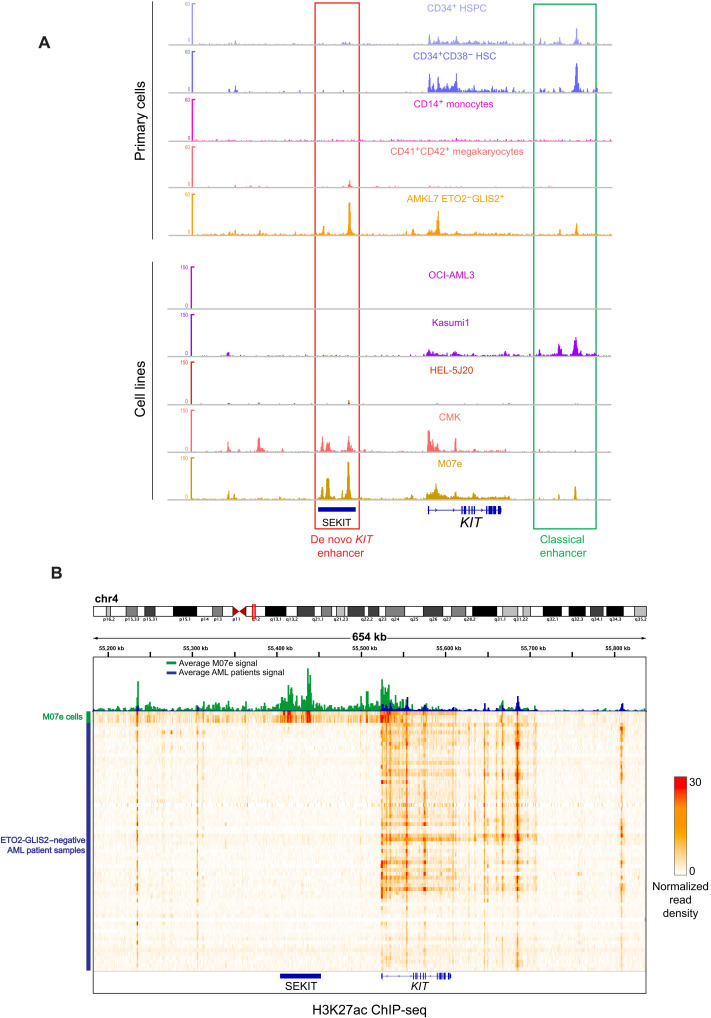
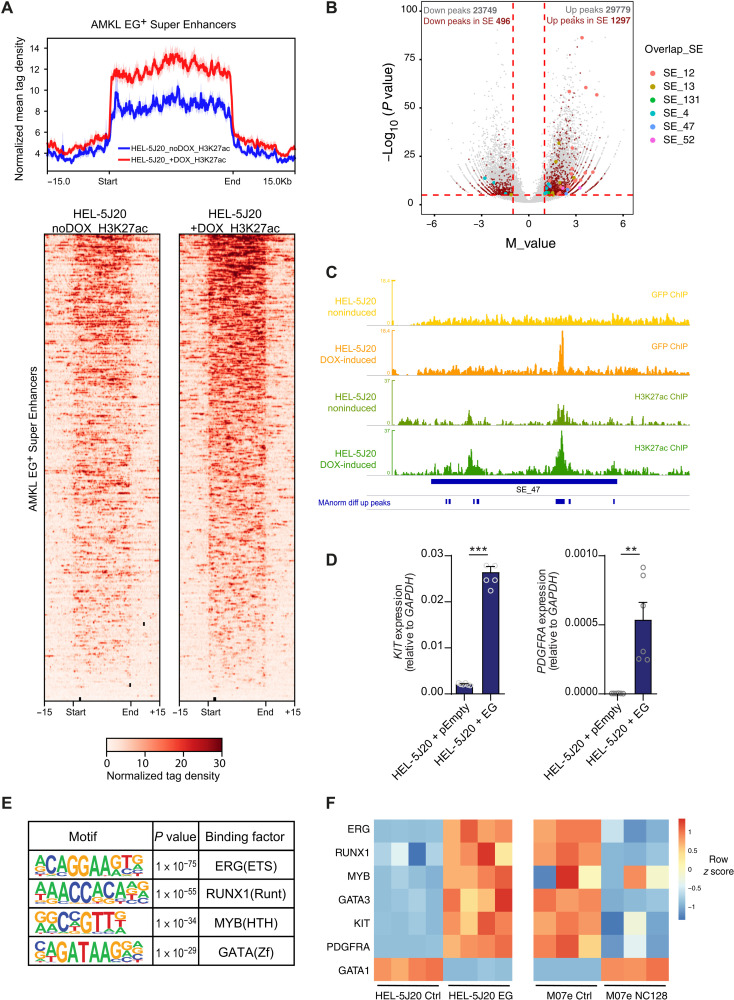
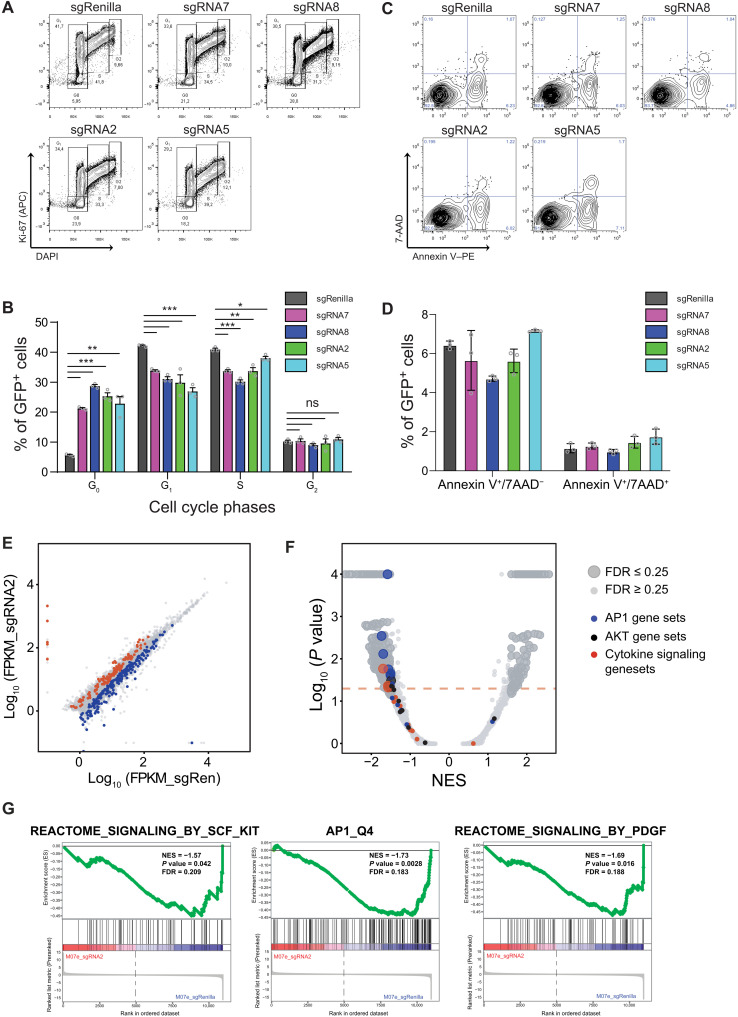
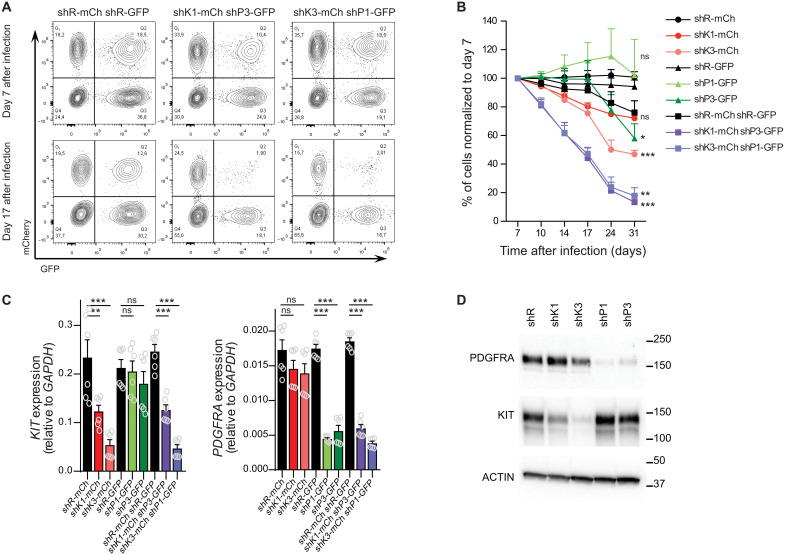
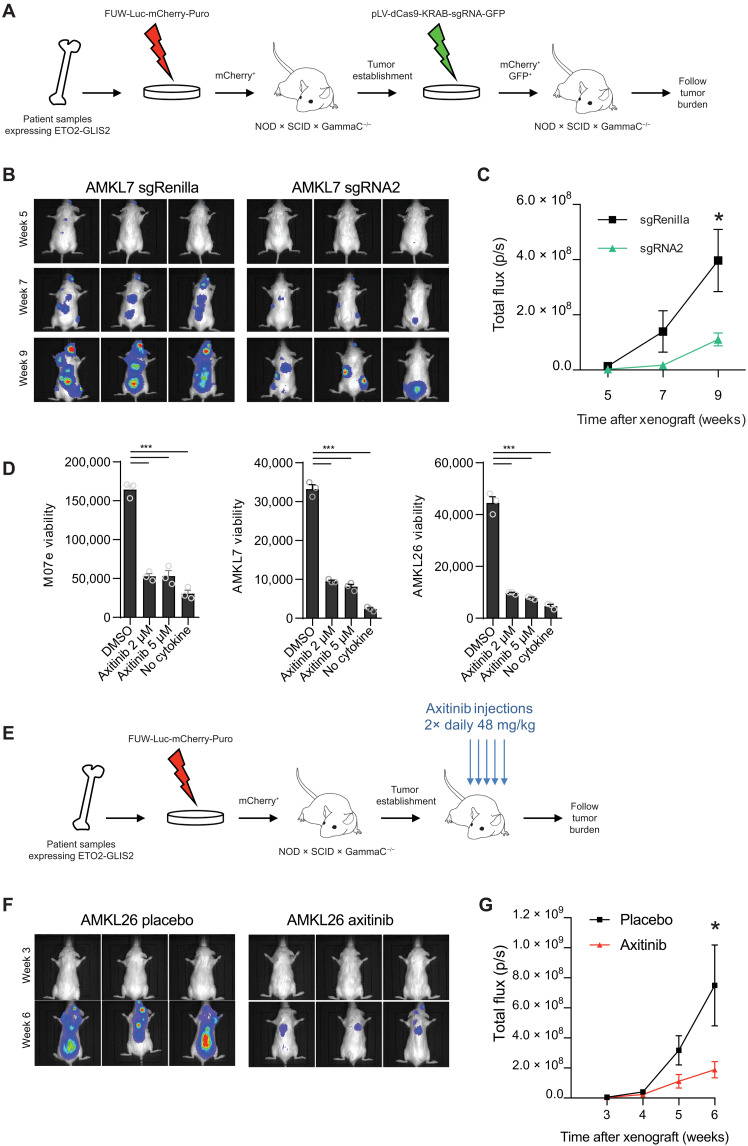
Super Enhancers (SEs) are clusters of regulatory elements associated with cell identity and disease. However, whether these elements are induced by oncogenes and can regulate gene modules cooperating for cancer cell transformation or maintenance remains elusive. To address this question, we conducted a genome-wide CRISPRi-based screening of SEs in ETO2-GLIS2+ acute megakaryoblastic leukemia. This approach revealed SEs essential for leukemic cell growth and survival that are induced by ETO2-GLIS2 expression. In particular, we identified a de novo SE specific of this leukemia subtype and regulating expression of tyrosine kinase-associated receptors KIT and PDGFRA. Combined expression of these two receptors was required for leukemic cell growth, and CRISPRi-mediated inhibition of this SE or treatment with tyrosine kinase inhibitors impaired progression of leukemia in vivo in patient-derived xenografts experiments. Our results show that fusion oncogenes, such as ETO2-GLIS2, can induce activation of SEs regulating essential gene modules synergizing for leukemia progression.
Figures
Similar articles
-
Developmental interplay between transcriptional alterations and a targetable cytokine signaling dependency in pediatric ETO2::GLIS2 leukemia.Mol Cancer. 2024 Sep 20;23(1):204. doi: 10.1186/s12943-024-02110-y. Mol Cancer. 2024. PMID: 39304903 Free PMC article.
-
Progressive chromatin rewiring by ETO2::GLIS2 revealed in a genome-edited human iPSC model of pediatric leukemia initiation.Blood. 2025 Apr 3;145(14):1510-1525. doi: 10.1182/blood.2024024505. Blood. 2025. PMID: 39656971
-
ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-Renewal in Pediatric Acute Megakaryoblastic Leukemia.Cancer Cell. 2017 Mar 13;31(3):452-465. doi: 10.1016/j.ccell.2017.02.006. Cancer Cell. 2017. PMID: 28292442
-
[Pediatric de novo acute megakaryoblastic leukemia: an affair of complexes].Med Sci (Paris). 2018 Nov;34(11):954-962. doi: 10.1051/medsci/2018237. Epub 2018 Dec 10. Med Sci (Paris). 2018. PMID: 30526836 Review. French.
-
[Research progress of super enhancer in cancer].Yi Chuan. 2019 Jan 20;41(1):41-51. doi: 10.16288/j.yczz.18-152. Yi Chuan. 2019. PMID: 30686784 Review. Chinese.
Cited by
-
Single-cell detection of primary transcripts, their genomic loci and nuclear factors by 3D immuno-RNA/DNA FISH in T cells.Front Immunol. 2023 May 4;14:1156077. doi: 10.3389/fimmu.2023.1156077. eCollection 2023. Front Immunol. 2023. PMID: 37215121 Free PMC article.
-
GLIS1-3: Links to Primary Cilium, Reprogramming, Stem Cell Renewal, and Disease.Cells. 2022 Jun 3;11(11):1833. doi: 10.3390/cells11111833. Cells. 2022. PMID: 35681527 Free PMC article. Review.
-
TAD border deletion at the Kit locus causes tissue-specific ectopic activation of a neighboring gene.Nat Commun. 2024 May 28;15(1):4521. doi: 10.1038/s41467-024-48523-7. Nat Commun. 2024. PMID: 38806452 Free PMC article.
-
PitViper: a software for comparative meta-analysis and annotation of functional screening data.NAR Genom Bioinform. 2024 May 25;6(2):lqae059. doi: 10.1093/nargab/lqae059. eCollection 2024 Jun. NAR Genom Bioinform. 2024. PMID: 38800827 Free PMC article.
-
CBFA2T3-GLIS2 mediates transcriptional regulation of developmental pathways through a gene regulatory network.Nat Commun. 2024 Oct 9;15(1):8747. doi: 10.1038/s41467-024-53158-9. Nat Commun. 2024. PMID: 39384814 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
