

Accounting for age of onset and family history improves power in genome-wide association studies
- PMID: 35139346
- PMCID: PMC8948165
- DOI: 10.1016/j.ajhg.2022.01.009
Accounting for age of onset and family history improves power in genome-wide association studies
Abstract
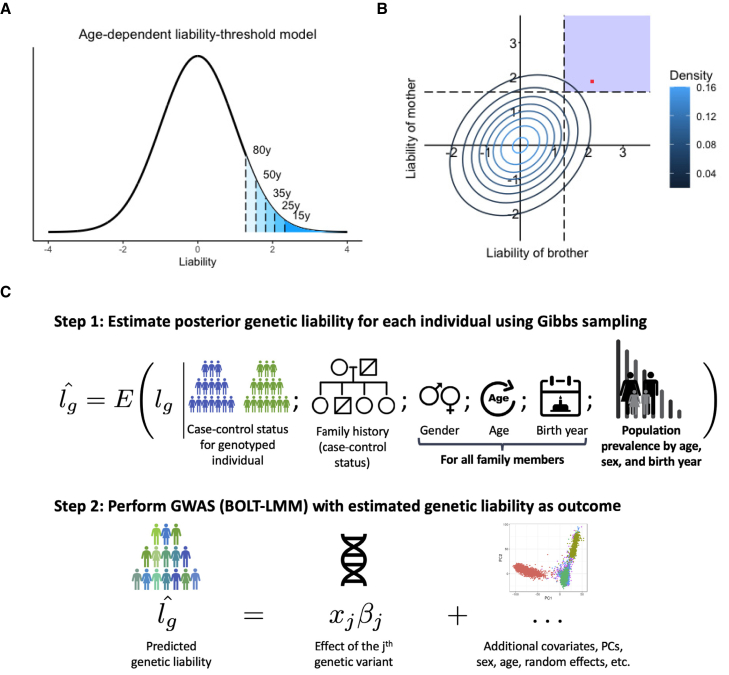
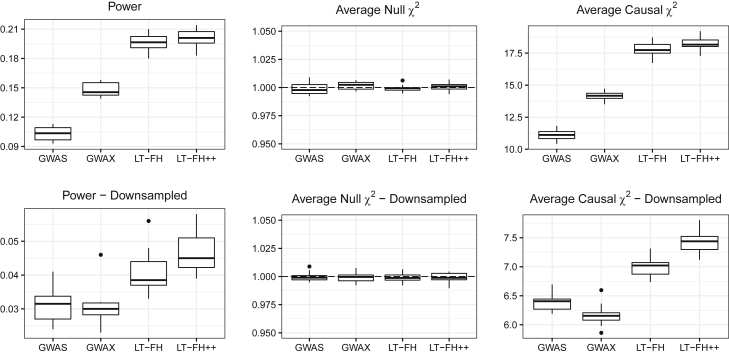
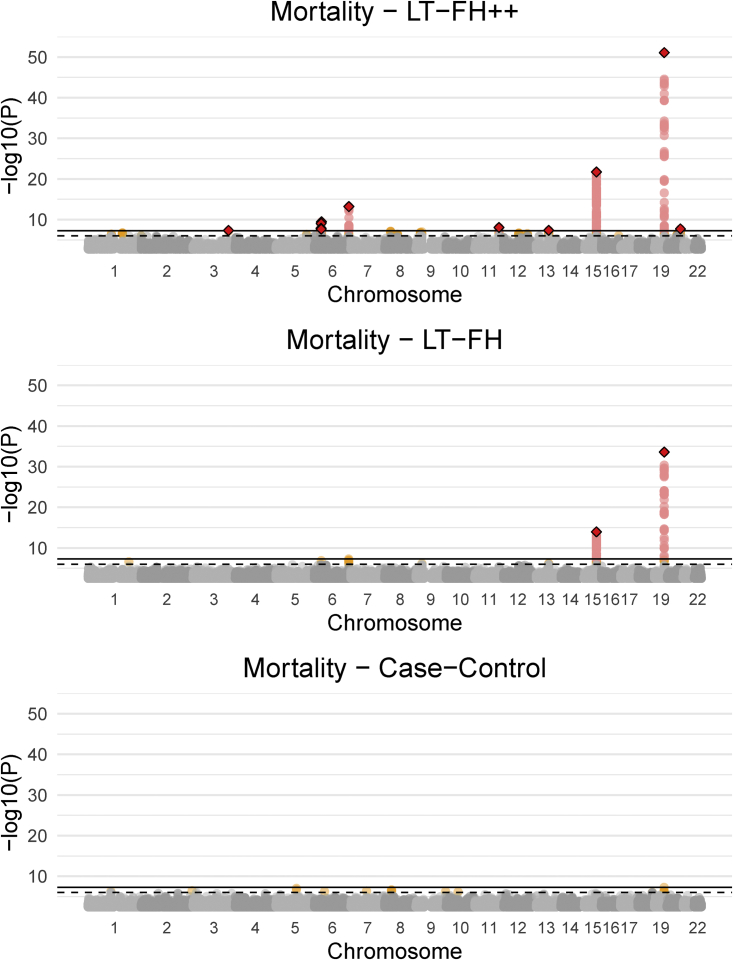
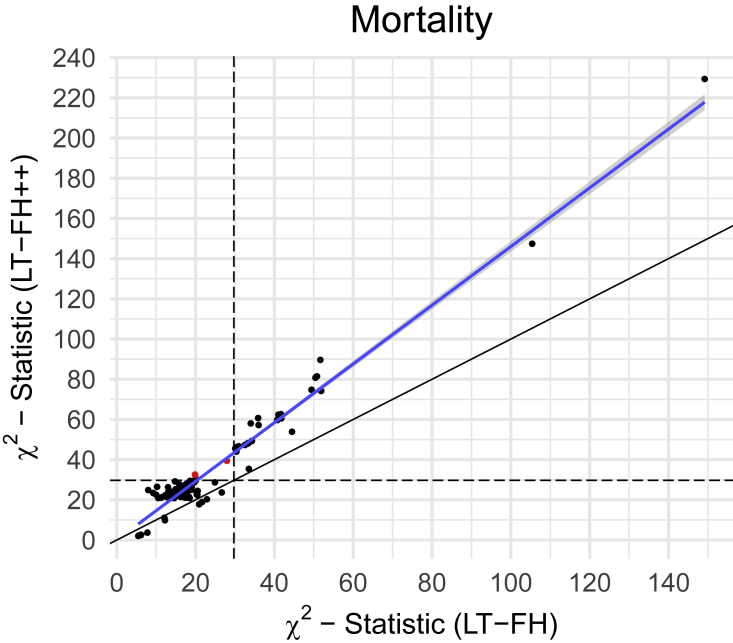
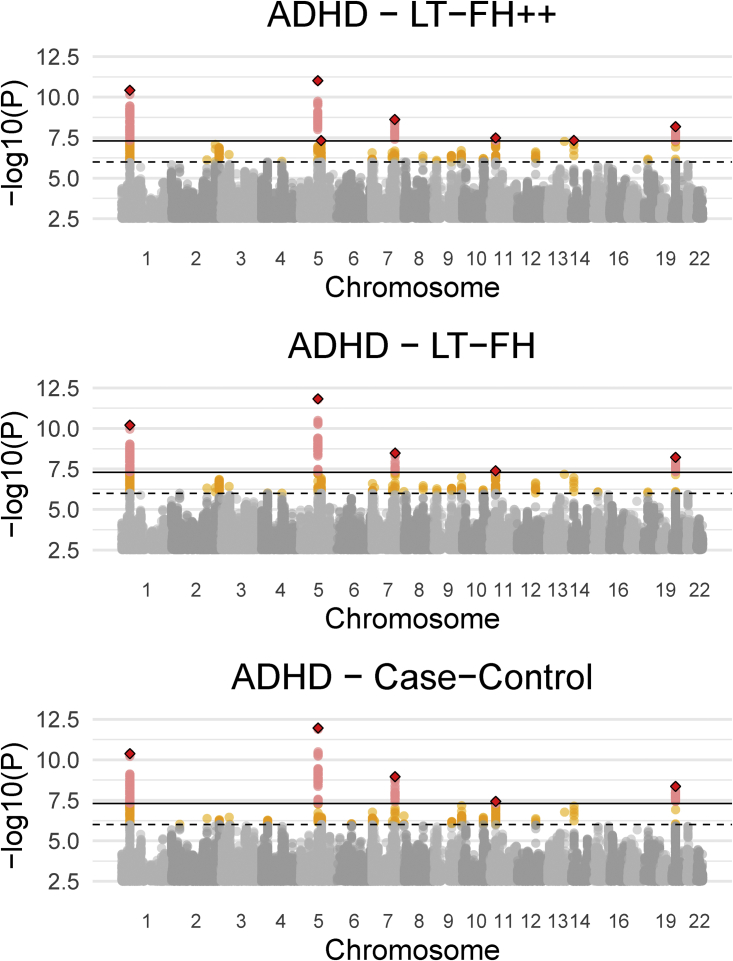
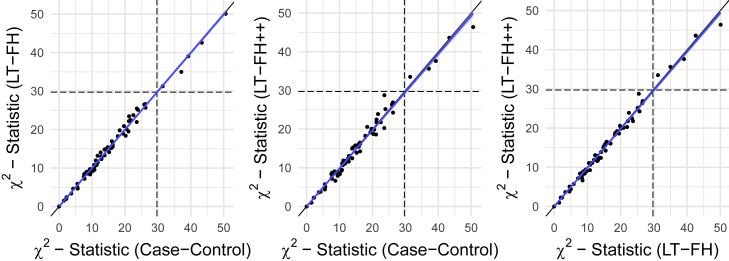
Genome-wide association studies (GWASs) have revolutionized human genetics, allowing researchers to identify thousands of disease-related genes and possible drug targets. However, case-control status does not account for the fact that not all controls may have lived through their period of risk for the disorder of interest. This can be quantified by examining the age-of-onset distribution and the age of the controls or the age of onset for cases. The age-of-onset distribution may also depend on information such as sex and birth year. In addition, family history is not routinely included in the assessment of control status. Here, we present LT-FH++, an extension of the liability threshold model conditioned on family history (LT-FH), which jointly accounts for age of onset and sex as well as family history. Using simulations, we show that, when family history and the age-of-onset distribution are available, the proposed approach yields statistically significant power gains over LT-FH and large power gains over genome-wide association study by proxy (GWAX). We applied our method to four psychiatric disorders available in the iPSYCH data and to mortality in the UK Biobank and found 20 genome-wide significant associations with LT-FH++, compared to ten for LT-FH and eight for a standard case-control GWAS. As more genetic data with linked electronic health records become available to researchers, we expect methods that account for additional health information, such as LT-FH++, to become even more beneficial.
Keywords: ADHD; LT-FH; LT-FH++; UKBB; age-of-onset; family history; genome-wide association study; iPSYCH; liability threshold model; mortality.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.C. has received honoraria for serving on the Scientific Advisory Board of Union Chimique Belge (UCB) Nordic and Eisai AB and for giving lectures for UCB Nordic and Eisai as well as travel funds from UCB Nordic and funding by the Novo Nordisk Foundation (grant number: NNF16OC0019126), the Central Denmark Region, and the Danish Epilepsy Association.
Figures
References
-
- Mahajan A., Taliun D., Thurner M., Robertson N.R., Torres J.M., Rayner N.W., Payne A.J., Steinthorsdottir V., Scott R.A., Grarup N., et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018;50:1505–1513. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
