

Selenophosphate synthetase 1 deficiency exacerbates osteoarthritis by dysregulating redox homeostasis
- PMID: 35140209
- PMCID: PMC8828855
- DOI: 10.1038/s41467-022-28385-7
Selenophosphate synthetase 1 deficiency exacerbates osteoarthritis by dysregulating redox homeostasis
Abstract
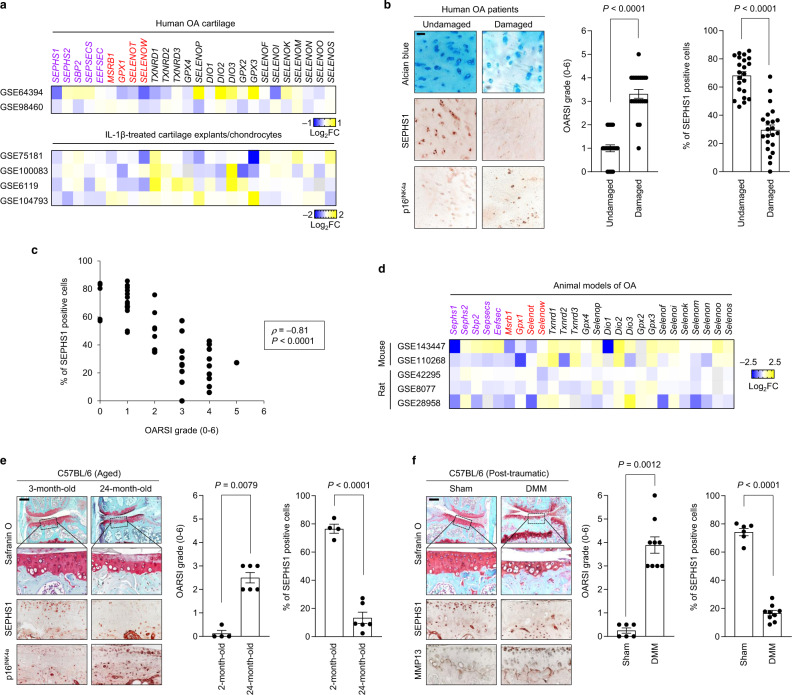
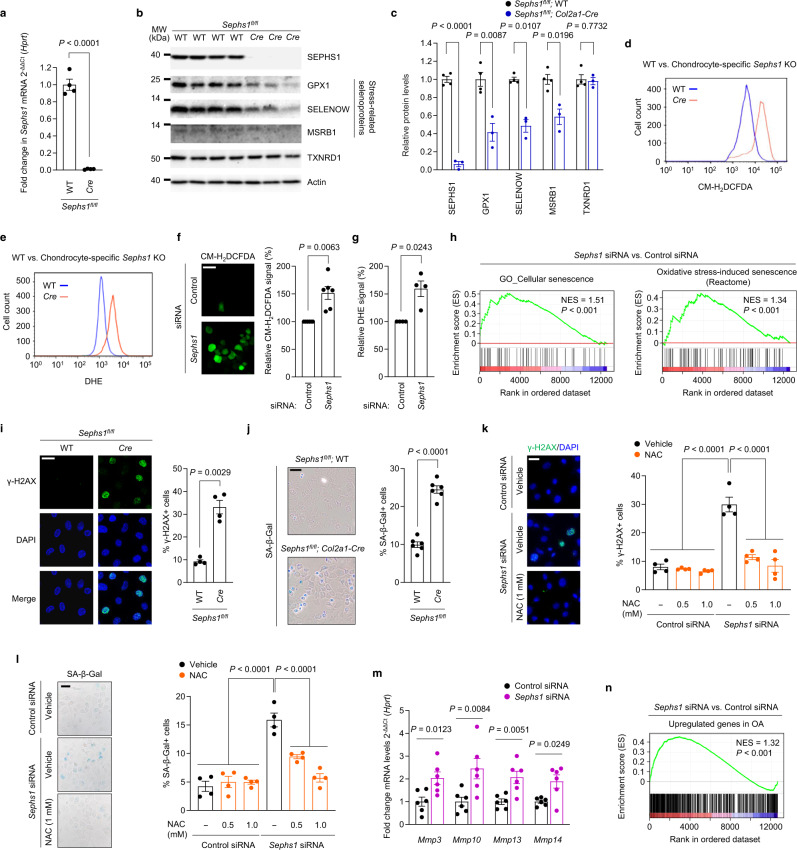
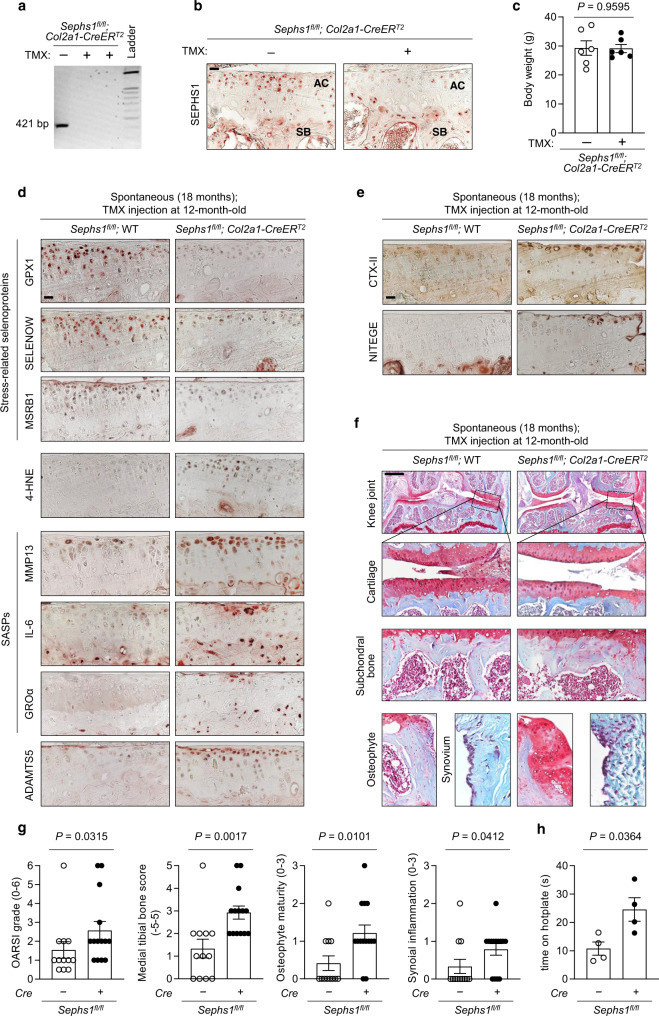
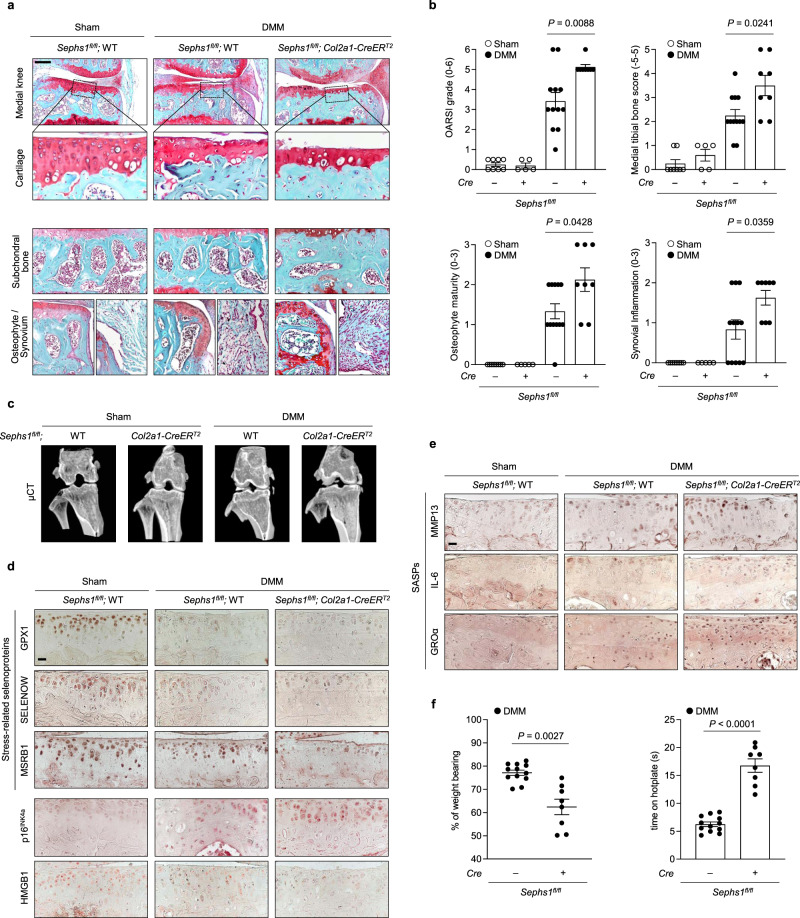
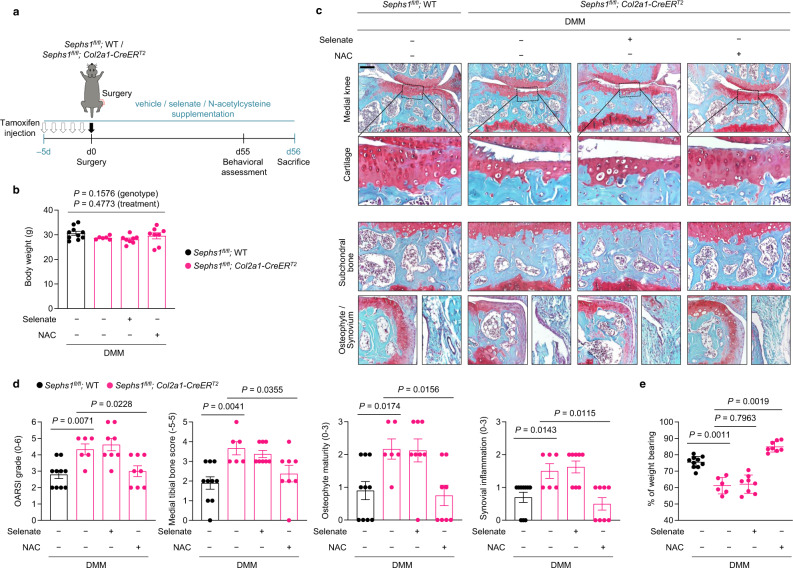
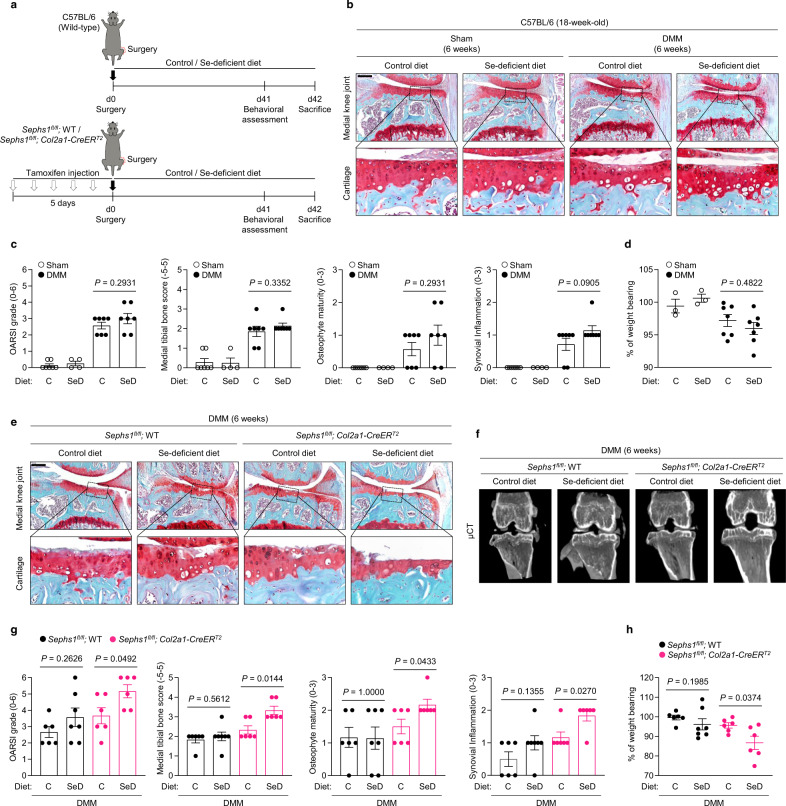
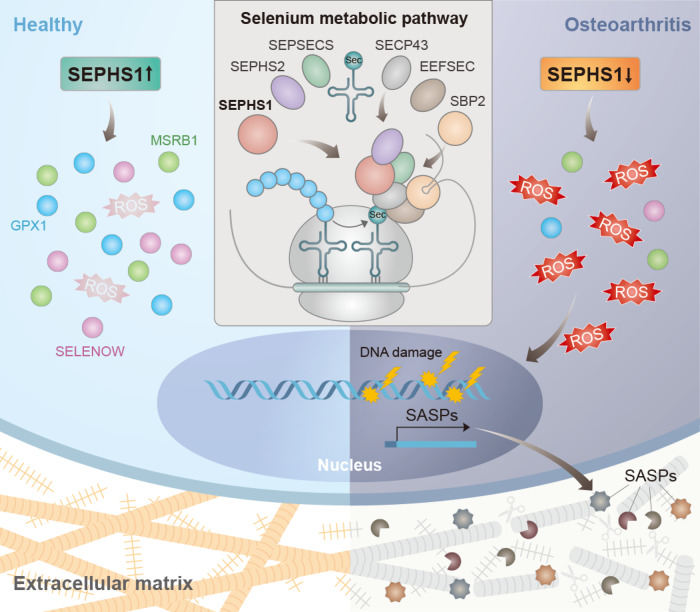
Aging and mechanical overload are prominent risk factors for osteoarthritis (OA), which lead to an imbalance in redox homeostasis. The resulting state of oxidative stress drives the pathological transition of chondrocytes during OA development. However, the specific molecular pathways involved in disrupting chondrocyte redox homeostasis remain unclear. Here, we show that selenophosphate synthetase 1 (SEPHS1) expression is downregulated in human and mouse OA cartilage. SEPHS1 downregulation impairs the cellular capacity to synthesize a class of selenoproteins with oxidoreductase functions in chondrocytes, thereby elevating the level of reactive oxygen species (ROS) and facilitating chondrocyte senescence. Cartilage-specific Sephs1 knockout in adult mice causes aging-associated OA, and augments post-traumatic OA, which is rescued by supplementation of N-acetylcysteine (NAC). Selenium-deficient feeding and Sephs1 knockout have synergistic effects in exacerbating OA pathogenesis in mice. Therefore, we propose that SEPHS1 is an essential regulator of selenium metabolism and redox homeostasis, and its dysregulation governs the progression of OA.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
SEPHS1: Its evolution, function and roles in development and diseases.Arch Biochem Biophys. 2022 Nov 15;730:109426. doi: 10.1016/j.abb.2022.109426. Epub 2022 Oct 4. Arch Biochem Biophys. 2022. PMID: 36202216 Free PMC article.
-
The role of selenium-mediated redox signaling by selenophosphate synthetase 1 (SEPHS1) in hESCs.Biochem Biophys Res Commun. 2019 Dec 3;520(2):406-412. doi: 10.1016/j.bbrc.2019.09.123. Epub 2019 Oct 11. Biochem Biophys Res Commun. 2019. PMID: 31607477
-
PDZK1 protects against mechanical overload-induced chondrocyte senescence and osteoarthritis by targeting mitochondrial function.Bone Res. 2024 Jul 17;12(1):41. doi: 10.1038/s41413-024-00344-6. Bone Res. 2024. PMID: 39019845 Free PMC article.
-
Reactive oxygen species, aging and articular cartilage homeostasis.Free Radic Biol Med. 2019 Feb 20;132:73-82. doi: 10.1016/j.freeradbiomed.2018.08.038. Epub 2018 Aug 31. Free Radic Biol Med. 2019. PMID: 30176344 Free PMC article. Review.
-
Selenophosphate synthetase 1 and its role in redox homeostasis, defense and proliferation.Free Radic Biol Med. 2018 Nov 1;127:190-197. doi: 10.1016/j.freeradbiomed.2018.04.577. Epub 2018 Apr 30. Free Radic Biol Med. 2018. PMID: 29715549 Review.
Cited by
-
Extracellular Vesicles Derived from Selenium-Deficient MAC-T Cells Aggravated Inflammation and Apoptosis by Triggering the Endoplasmic Reticulum (ER) Stress/PI3K-AKT-mTOR Pathway in Bovine Mammary Epithelial Cells.Antioxidants (Basel). 2023 Dec 5;12(12):2077. doi: 10.3390/antiox12122077. Antioxidants (Basel). 2023. PMID: 38136197 Free PMC article.
-
Cartilage-targeting peptide-modified cerium oxide nanoparticles alleviate oxidative stress and cartilage damage in osteoarthritis.J Nanobiotechnology. 2024 Dec 19;22(1):784. doi: 10.1186/s12951-024-03068-1. J Nanobiotechnology. 2024. PMID: 39702137 Free PMC article.
-
Unveiling the Role of Selenium in Child Development: Impacts on Growth, Neurodevelopment and Immunity.J Clin Med. 2025 Feb 14;14(4):1274. doi: 10.3390/jcm14041274. J Clin Med. 2025. PMID: 40004804 Free PMC article. Review.
-
Therapeutic prospects and potential mechanisms of Prdx6: as a novel target in musculoskeletal disorders.Front Physiol. 2025 Apr 17;16:1524100. doi: 10.3389/fphys.2025.1524100. eCollection 2025. Front Physiol. 2025. PMID: 40313876 Free PMC article. Review.
-
SEPHS1: Its evolution, function and roles in development and diseases.Arch Biochem Biophys. 2022 Nov 15;730:109426. doi: 10.1016/j.abb.2022.109426. Epub 2022 Oct 4. Arch Biochem Biophys. 2022. PMID: 36202216 Free PMC article.
References
-
- Little CB, Hunter DJ. Post-traumatic osteoarthritis: from mouse models to clinical trials. Nat. Rev. Rheumatol. 2013;9:485–497. - PubMed
-
- Muir H. The chondrocyte, architect of cartilage. Biomechanics, structure, function, and molecular biology of cartilage matrix macromolecules. Bioessays. 1995;17:1039–1048. - PubMed
-
- Mobasheri A, et al. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017;13:302–311. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
