

Caffeine blocks SREBP2-induced hepatic PCSK9 expression to enhance LDLR-mediated cholesterol clearance
- PMID: 35140212
- PMCID: PMC8828868
- DOI: 10.1038/s41467-022-28240-9
Caffeine blocks SREBP2-induced hepatic PCSK9 expression to enhance LDLR-mediated cholesterol clearance
Abstract
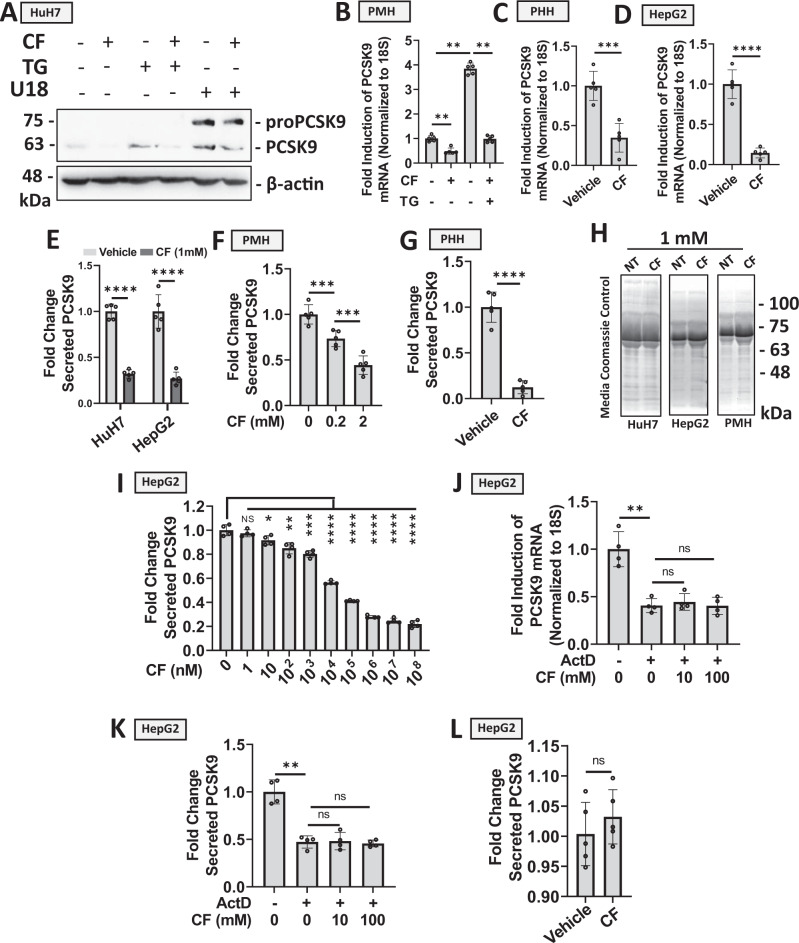
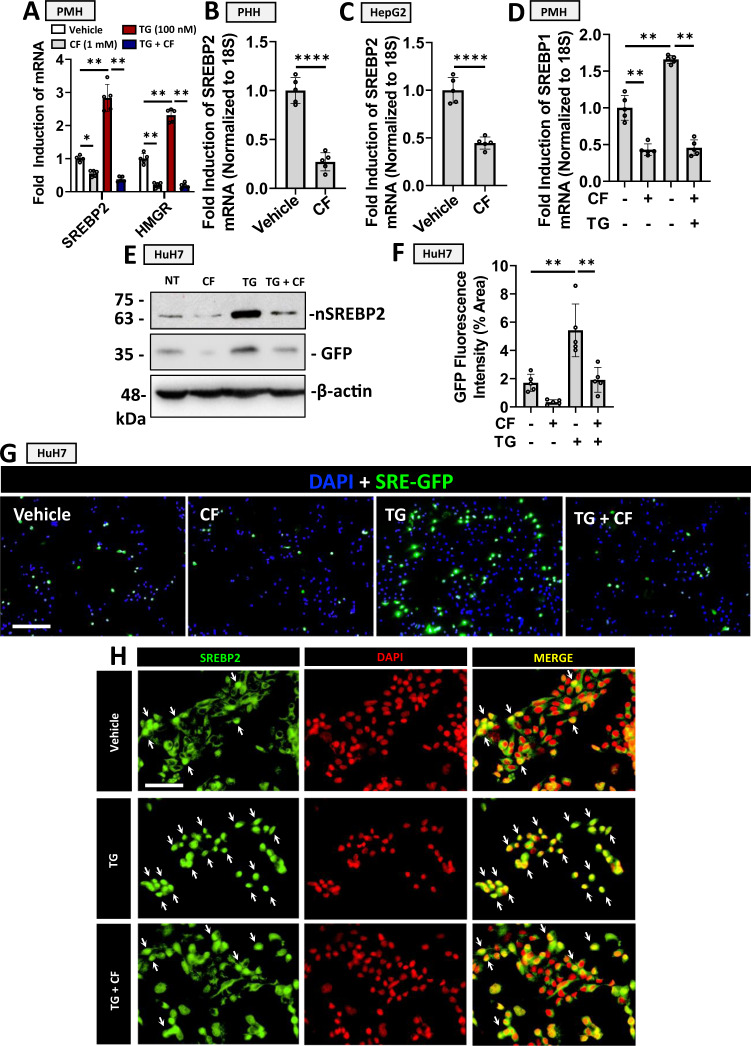
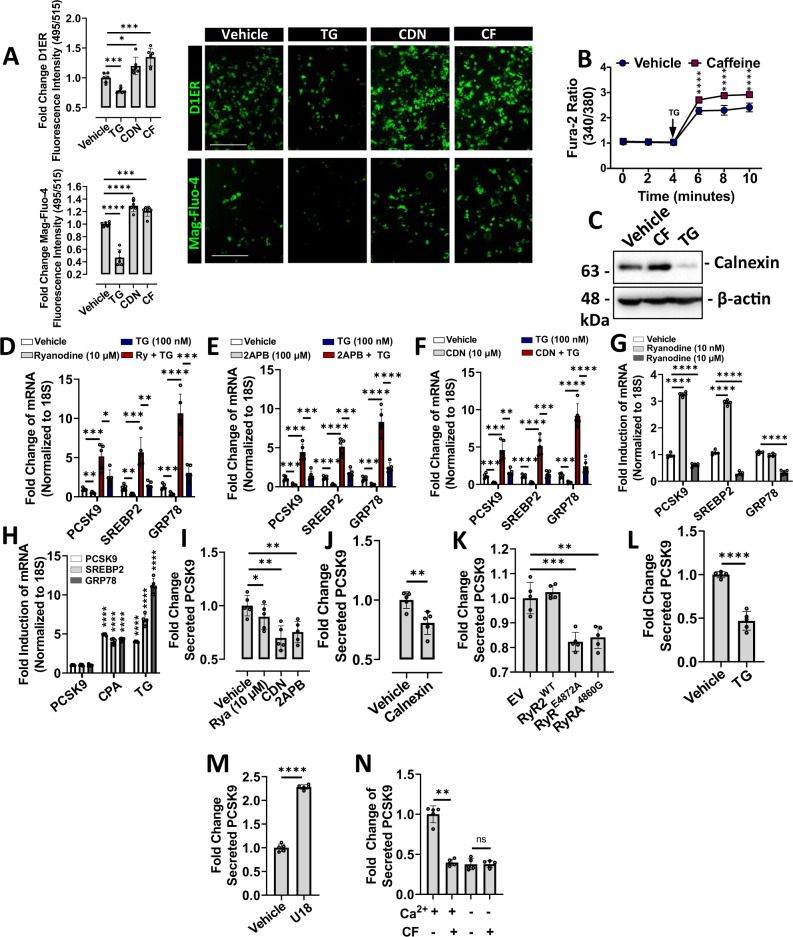
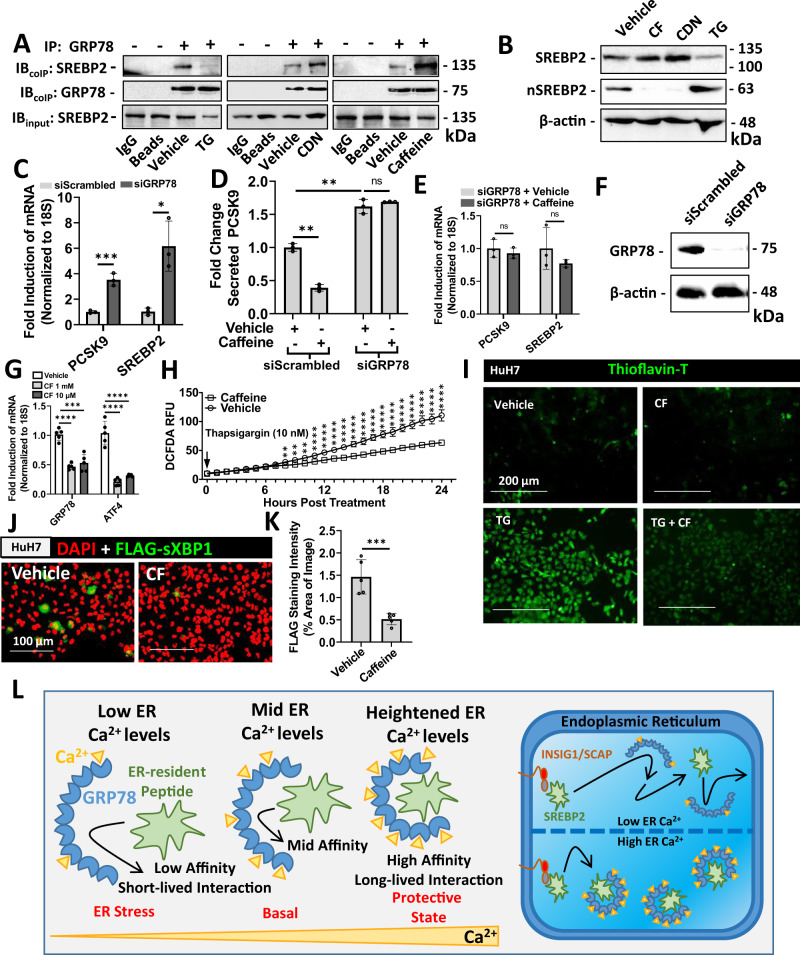
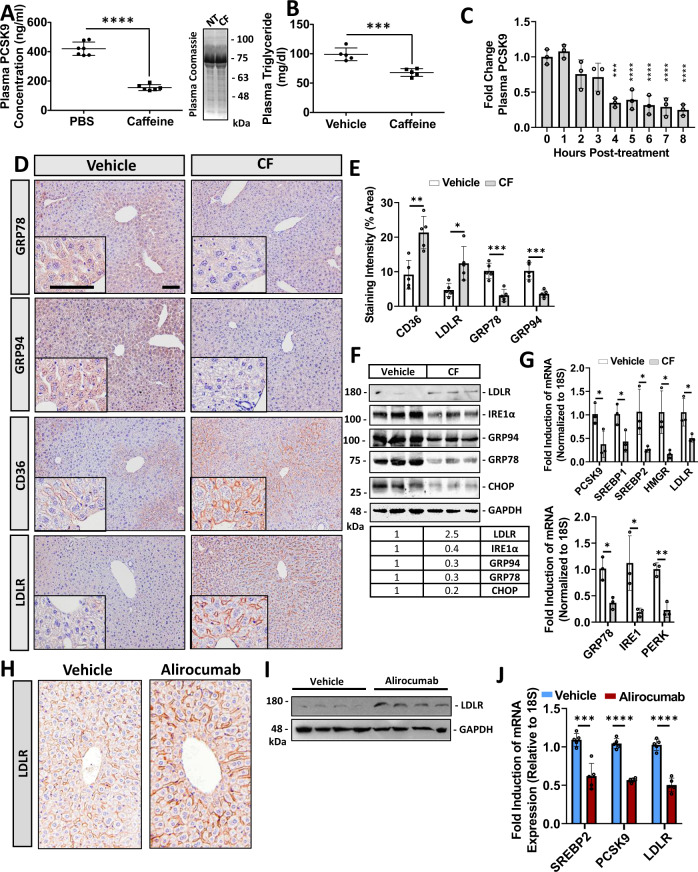
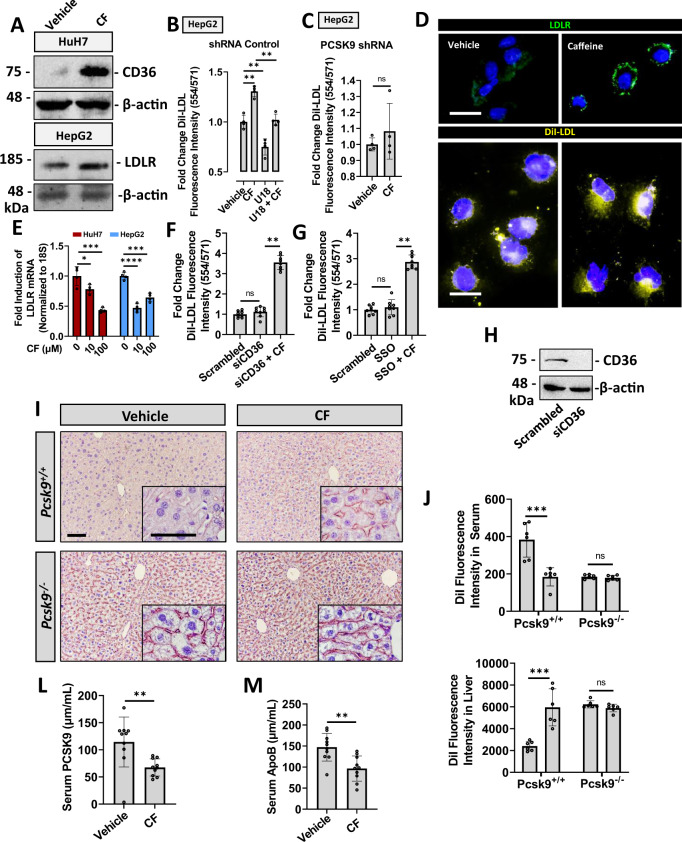
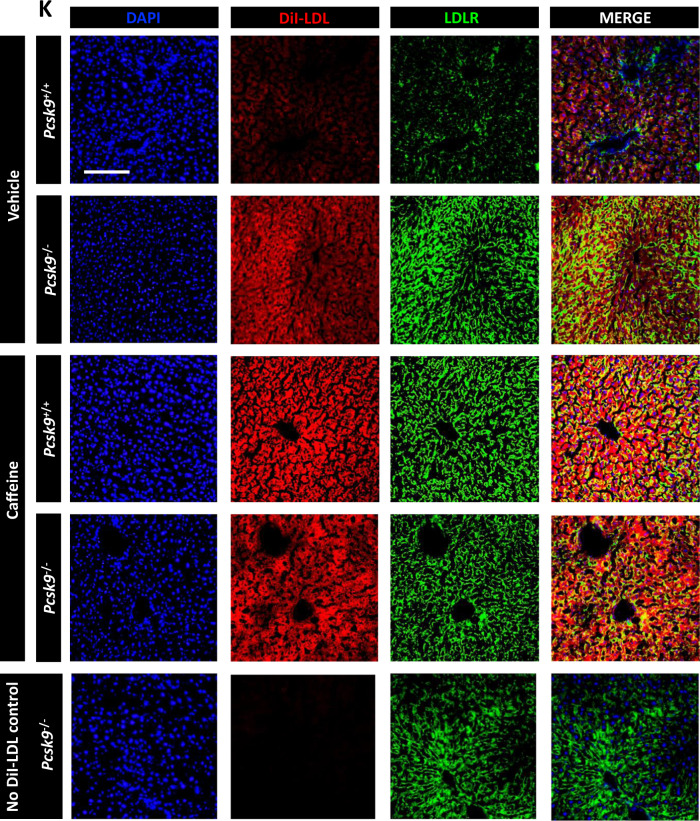
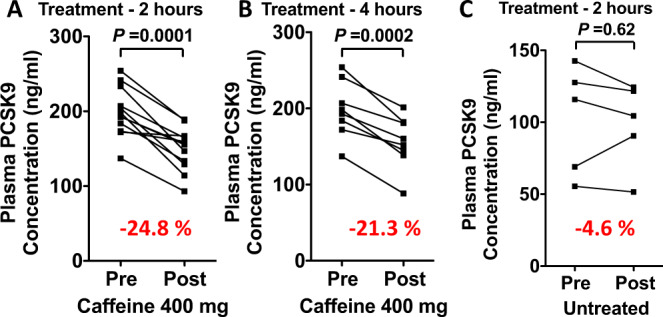
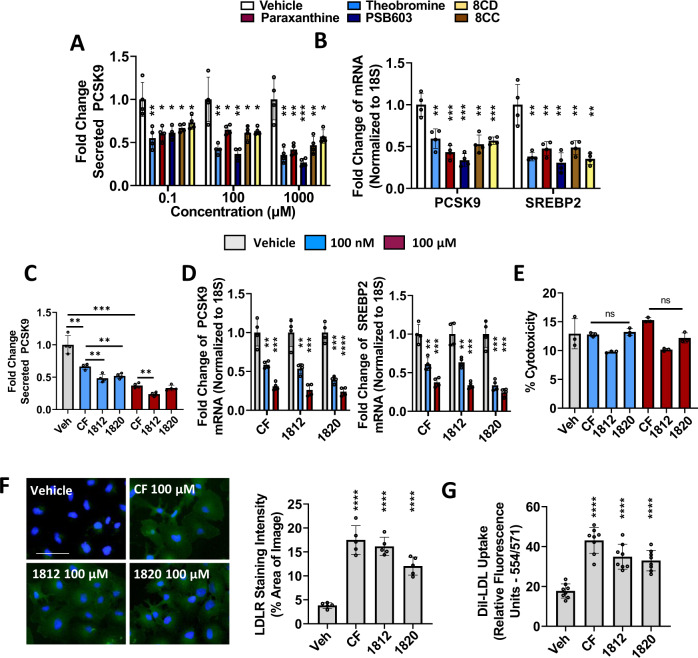
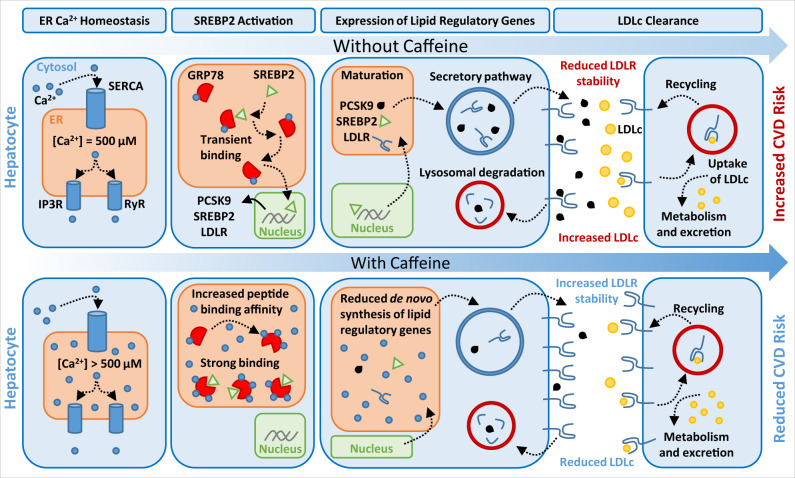
Evidence suggests that caffeine (CF) reduces cardiovascular disease (CVD) risk. However, the mechanism by which this occurs has not yet been uncovered. Here, we investigated the effect of CF on the expression of two bona fide regulators of circulating low-density lipoprotein cholesterol (LDLc) levels; the proprotein convertase subtilisin/kexin type 9 (PCSK9) and the low-density lipoprotein receptor (LDLR). Following the observation that CF reduced circulating PCSK9 levels and increased hepatic LDLR expression, additional CF-derived analogs with increased potency for PCSK9 inhibition compared to CF itself were developed. The PCSK9-lowering effect of CF was subsequently confirmed in a cohort of healthy volunteers. Mechanistically, we demonstrate that CF increases hepatic endoplasmic reticulum (ER) Ca2+ levels to block transcriptional activation of the sterol regulatory element-binding protein 2 (SREBP2) responsible for the regulation of PCSK9, thereby increasing the expression of the LDLR and clearance of LDLc. Our findings highlight ER Ca2+ as a master regulator of cholesterol metabolism and identify a mechanism by which CF may protect against CVD.
© 2022. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: P.F.L., J.H.B, P.S., M.S., J.M., and R.C.A. have filed an institution-owned patent, entitled: “Compounds for Reducing Cholesterol and Treating Liver and Kidney Disease” (18-069_USProv) that relates to the development of caffeine and methylxanthine derivatives to lower cholesterol. R.C.A, J.M., G.P., and M.A.T. have an equity stake in Systemic Therapeutics. G.P. has received honoraria from Amgen and Sanofi. G.R.S. is a co-founder and shareholder of Espervita Therapeutics, a company developing new medications for liver cancer. McMaster University has received funding from Espervita Therapeutics, Esperion Therapeutics, Poxel Pharmaceuticals and Novo Nordisk for research conducted in the laboratory of G.R.S. G.R.S. has received consulting/speaking fees from Astra Zeneca, Eli Lilly, Esperion Therapeutics, Merck, Poxel Pharmaceuticals and Takeda. M.A.T. is the CEO of Exerkine Corporation and Stayabove Nutrition and is actively evaluating and marketing multi-nutrient supplements for the treatment of aging, obesity, muscular dystrophy and mitochondrial disease. No other authors declare any competing interests.
Figures
References
-
- Abifadel M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34:154–156. - PubMed
-
- Benjannet S, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 2004;279:48865–48875. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
