

Signatures of TOP1 transcription-associated mutagenesis in cancer and germline
- PMID: 35140396
- PMCID: PMC8866115
- DOI: 10.1038/s41586-022-04403-y
Signatures of TOP1 transcription-associated mutagenesis in cancer and germline
Erratum in
-
Publisher Correction: Signatures of TOP1 transcription-associated mutagenesis in cancer and germline.Nature. 2022 May;605(7910):E7. doi: 10.1038/s41586-022-04812-z. Nature. 2022. PMID: 35504971 Free PMC article. No abstract available.
Abstract
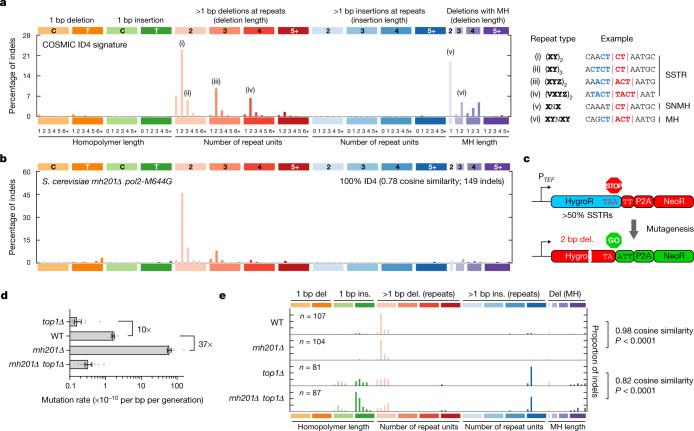
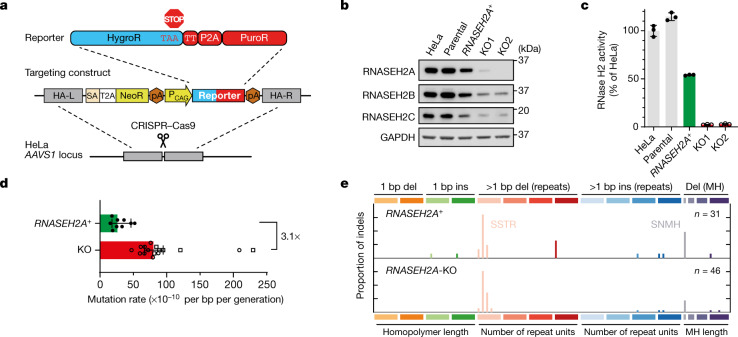
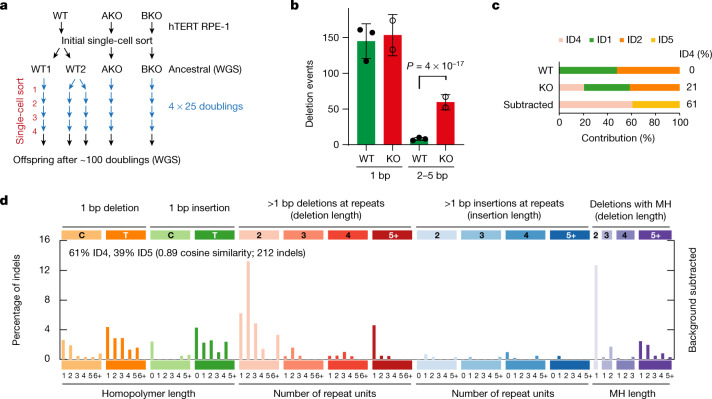
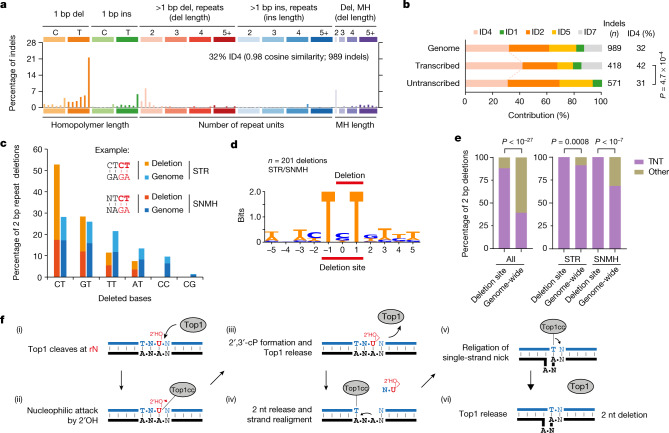
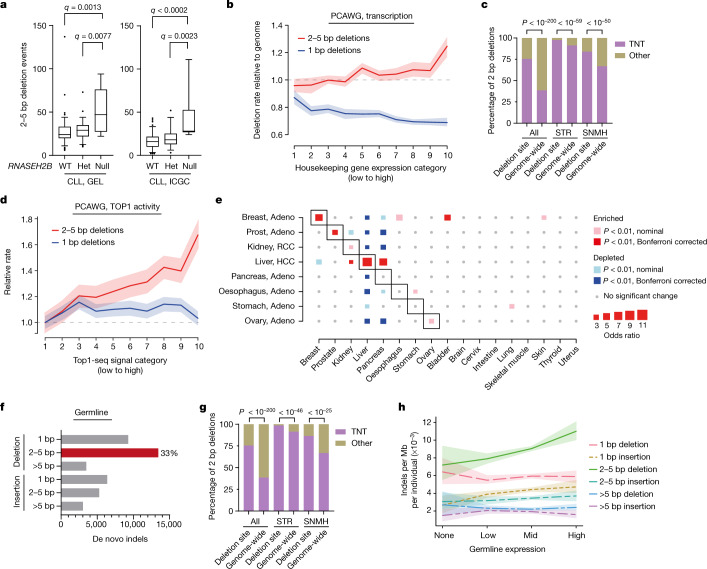
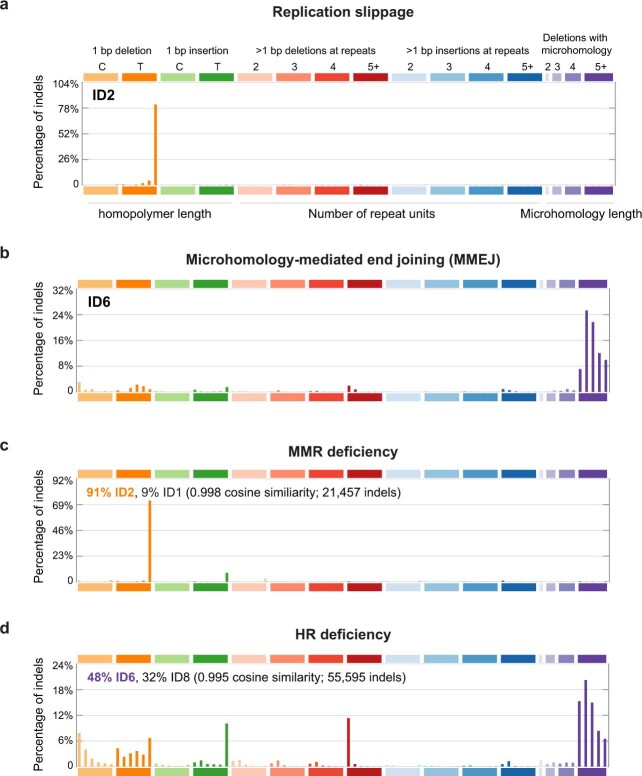
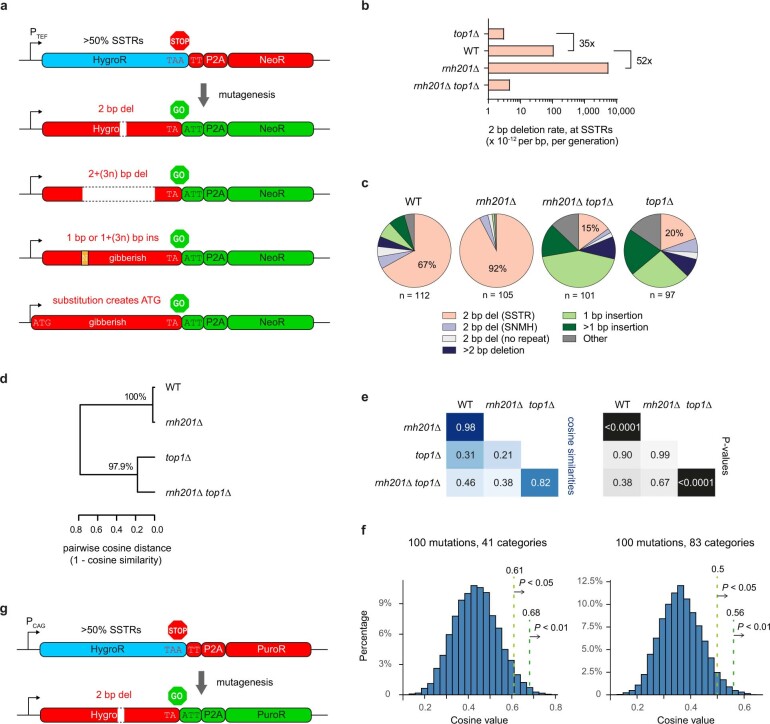
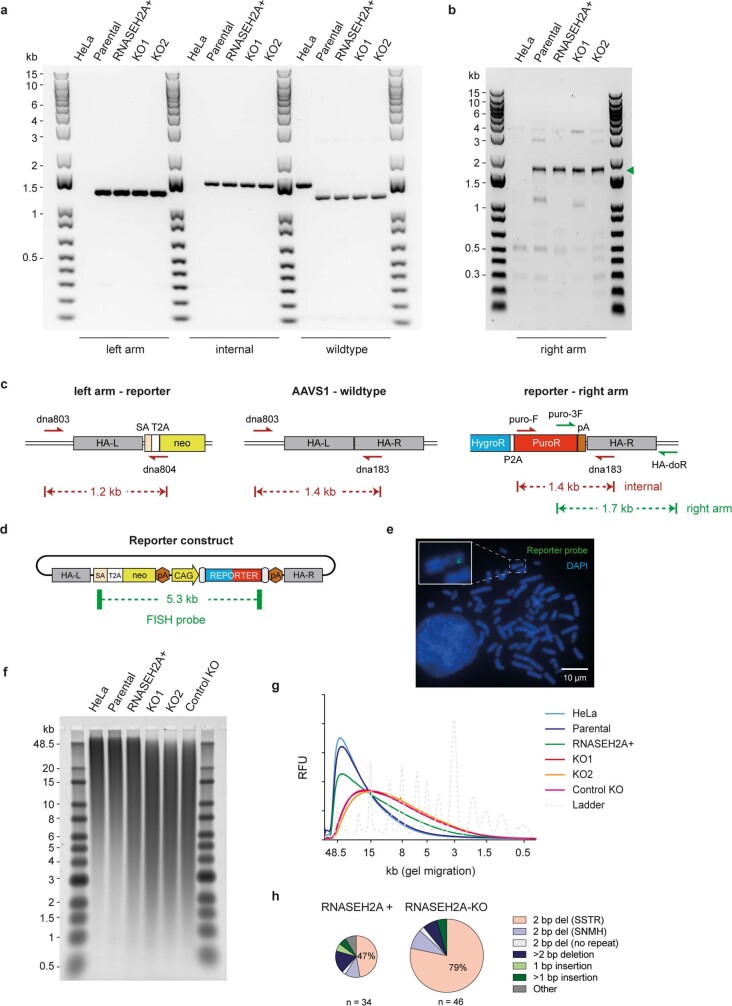
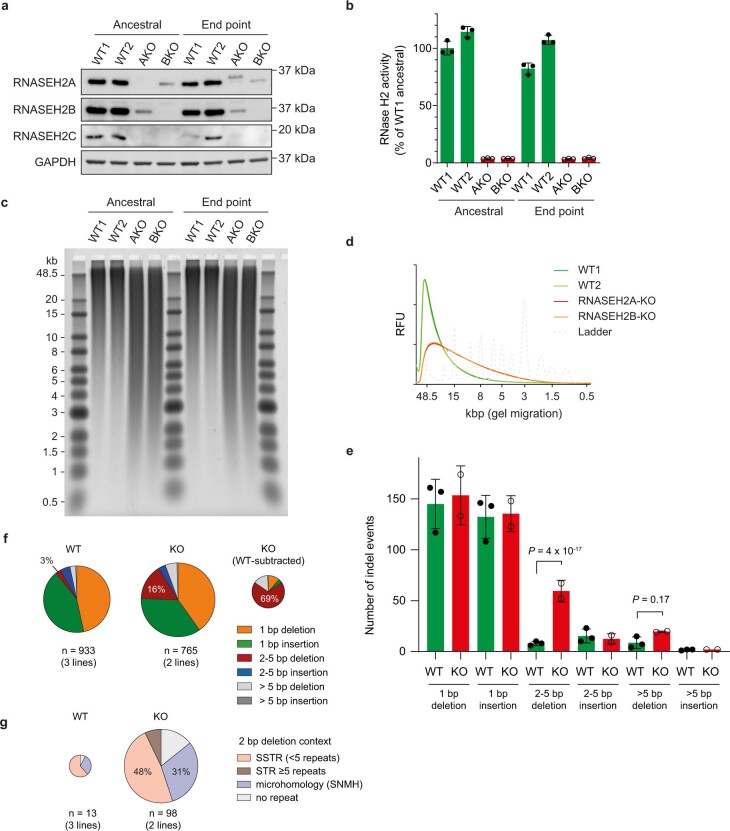
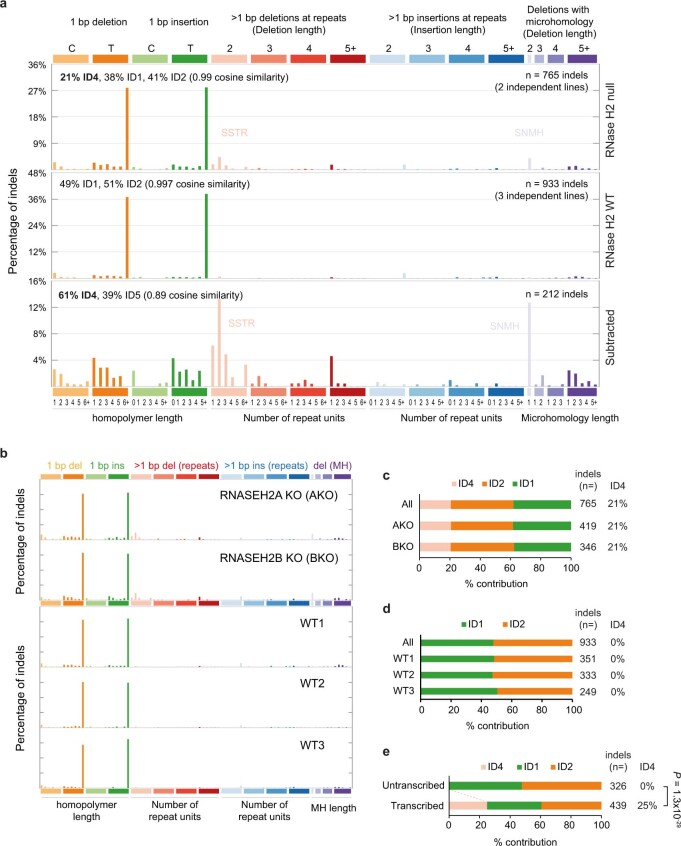
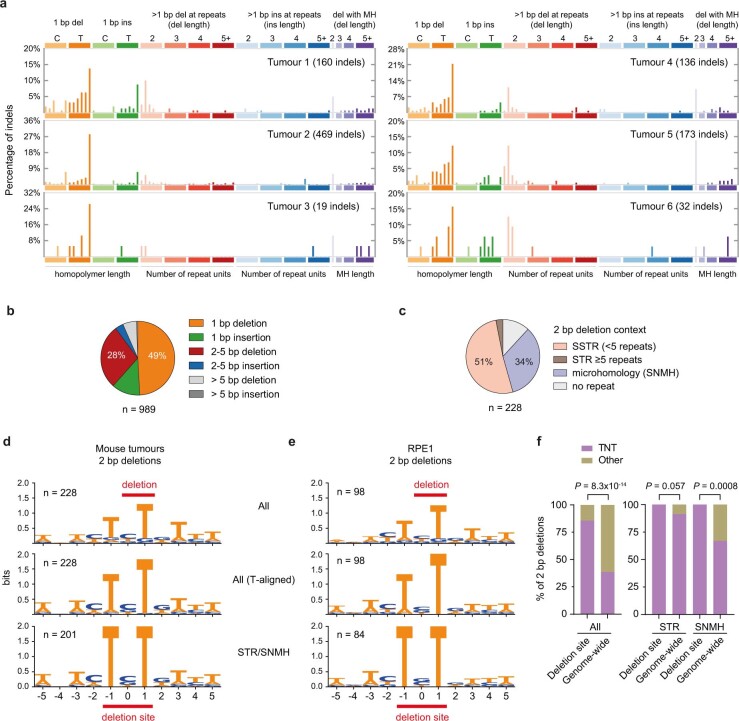
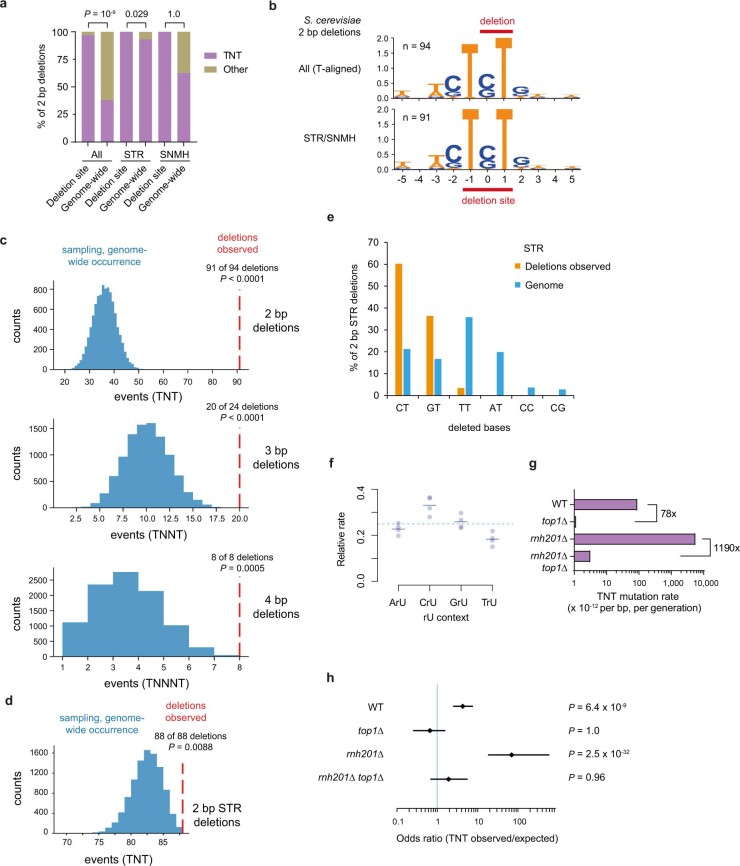
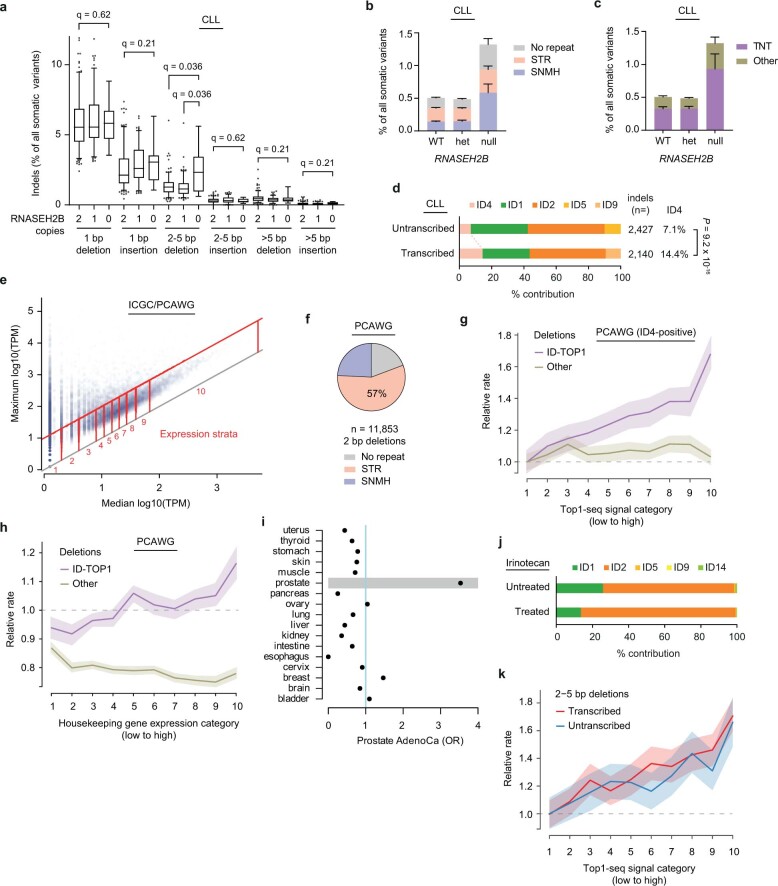
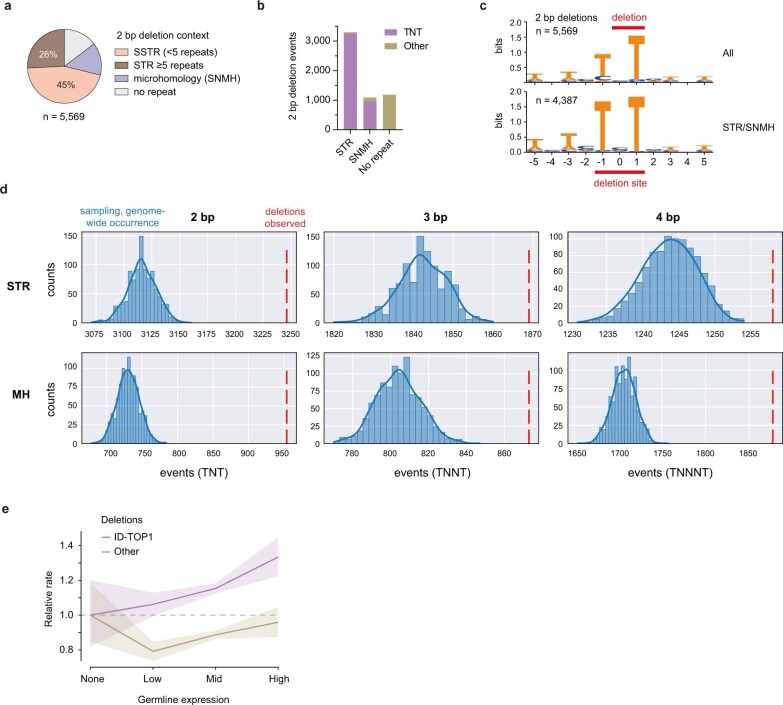
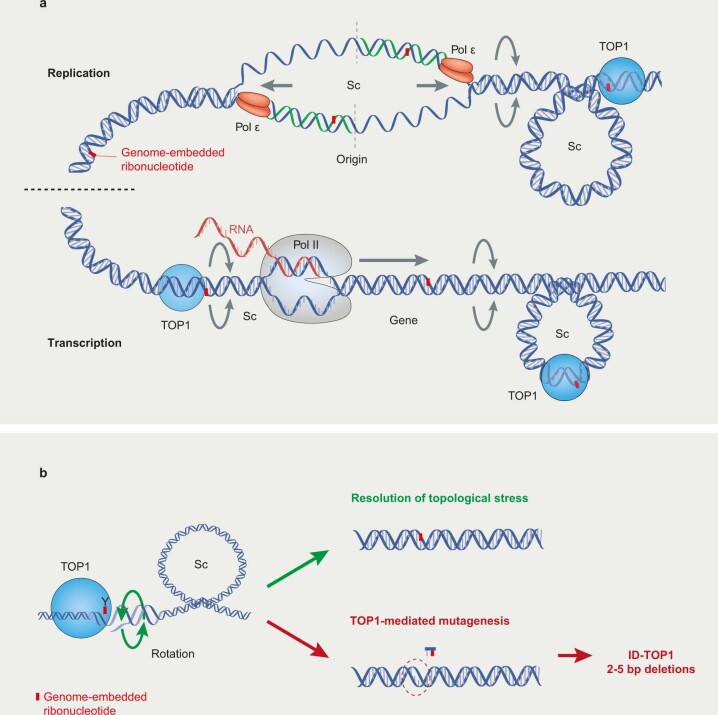
The mutational landscape is shaped by many processes. Genic regions are vulnerable to mutation but are preferentially protected by transcription-coupled repair1. In microorganisms, transcription has been demonstrated to be mutagenic2,3; however, the impact of transcription-associated mutagenesis remains to be established in higher eukaryotes4. Here we show that ID4-a cancer insertion-deletion (indel) mutation signature of unknown aetiology5 characterized by short (2 to 5 base pair) deletions -is due to a transcription-associated mutagenesis process. We demonstrate that defective ribonucleotide excision repair in mammals is associated with the ID4 signature, with mutations occurring at a TNT sequence motif, implicating topoisomerase 1 (TOP1) activity at sites of genome-embedded ribonucleotides as a mechanistic basis. Such TOP1-mediated deletions occur somatically in cancer, and the ID-TOP1 signature is also found in physiological settings, contributing to genic de novo indel mutations in the germline. Thus, although topoisomerases protect against genome instability by relieving topological stress6, their activity may also be an important source of mutations in the human genome.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Unwinding the mutational signatures of a DNA topoisomerase enzyme.Nature. 2022 Feb;602(7898):580-581. doi: 10.1038/d41586-022-00301-5. Nature. 2022. PMID: 35140368 No abstract available.
References
-
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. - PubMed
-
- Datta A, Jinks-Robertson S. Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science. 1995;268:1616–1619. - PubMed
MeSH terms
Substances
Grants and funding
- MR/K001744/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00007/11/MRC_/Medical Research Council/United Kingdom
- 28640/CRUK_/Cancer Research UK/United Kingdom
- MC_EX_MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00007/5/MRC_/Medical Research Council/United Kingdom
- 27589/CRUK_/Cancer Research UK/United Kingdom
- 25514/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_00009/2/MRC_/Medical Research Council/United Kingdom
- MC_PC_14089/MRC_/Medical Research Council/United Kingdom
- 19771/CRUK_/Cancer Research UK/United Kingdom
- 204802/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- 27963/CRUK_/Cancer Research UK/United Kingdom
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
