

Mapping clustered mutations in cancer reveals APOBEC3 mutagenesis of ecDNA
- PMID: 35140399
- PMCID: PMC8850194
- DOI: 10.1038/s41586-022-04398-6
Mapping clustered mutations in cancer reveals APOBEC3 mutagenesis of ecDNA
Abstract
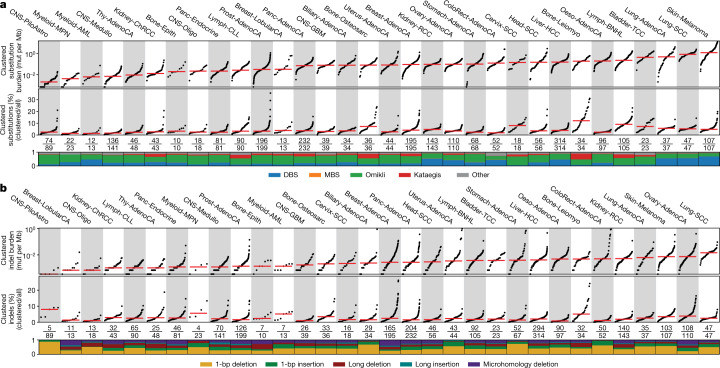
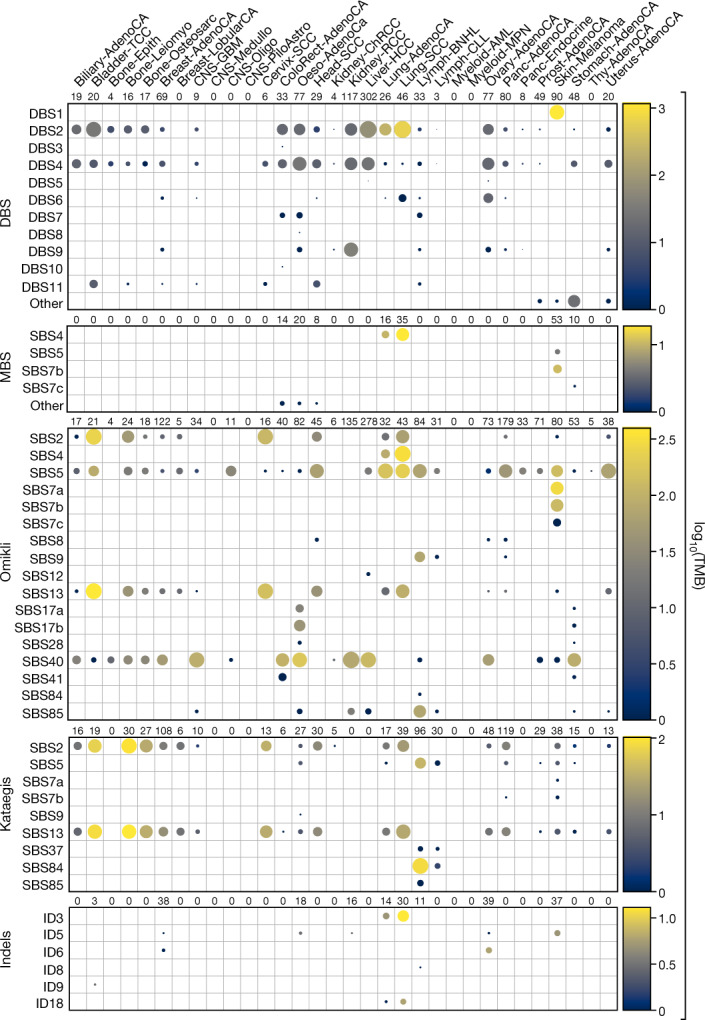
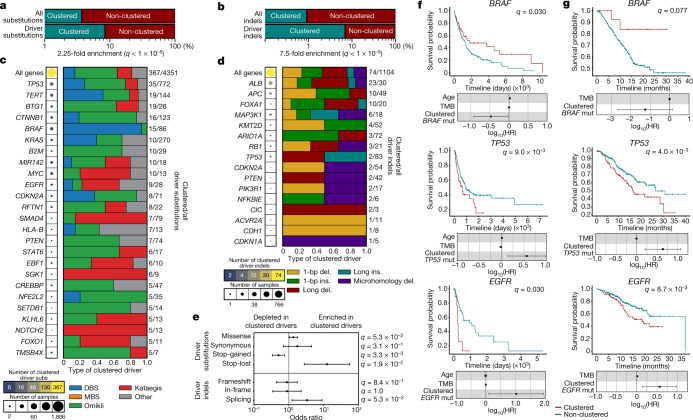
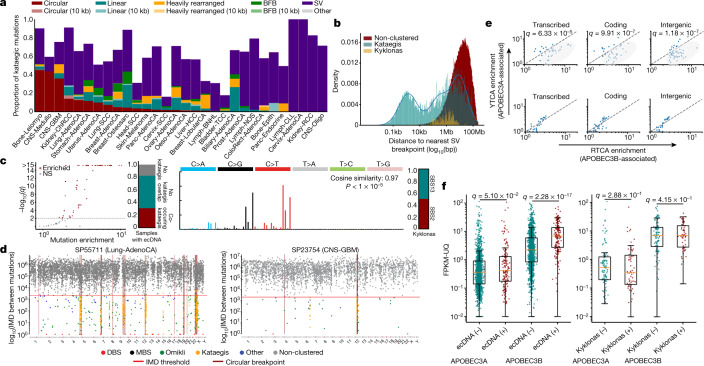
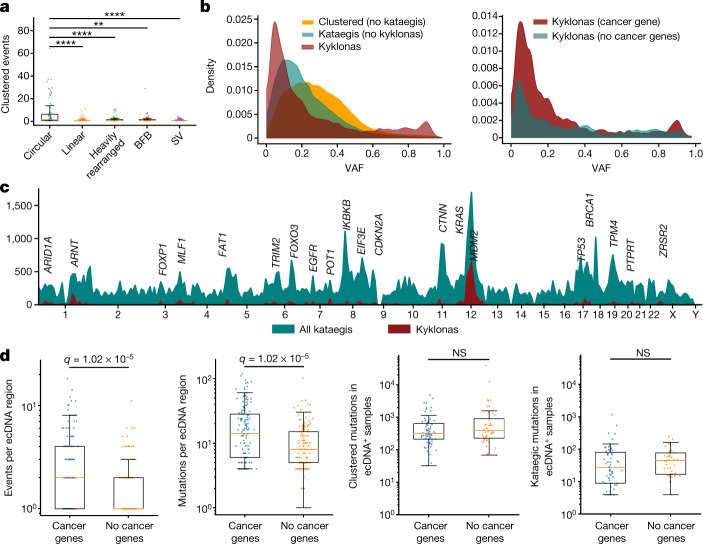
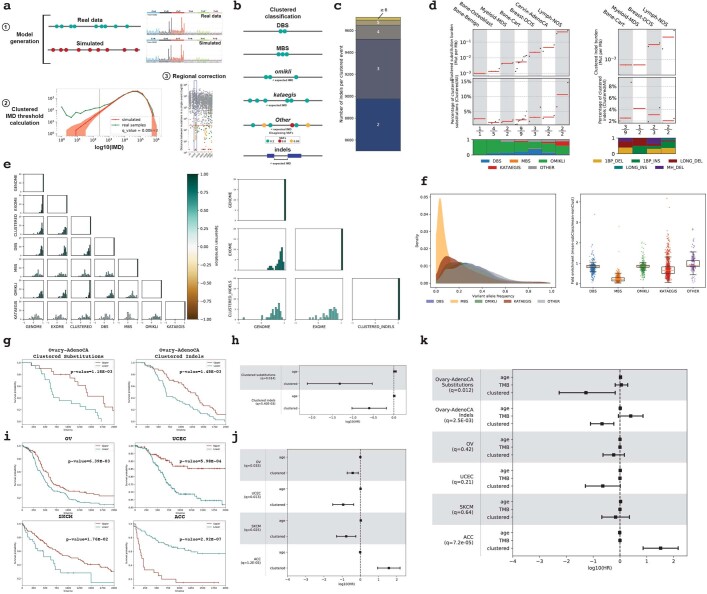
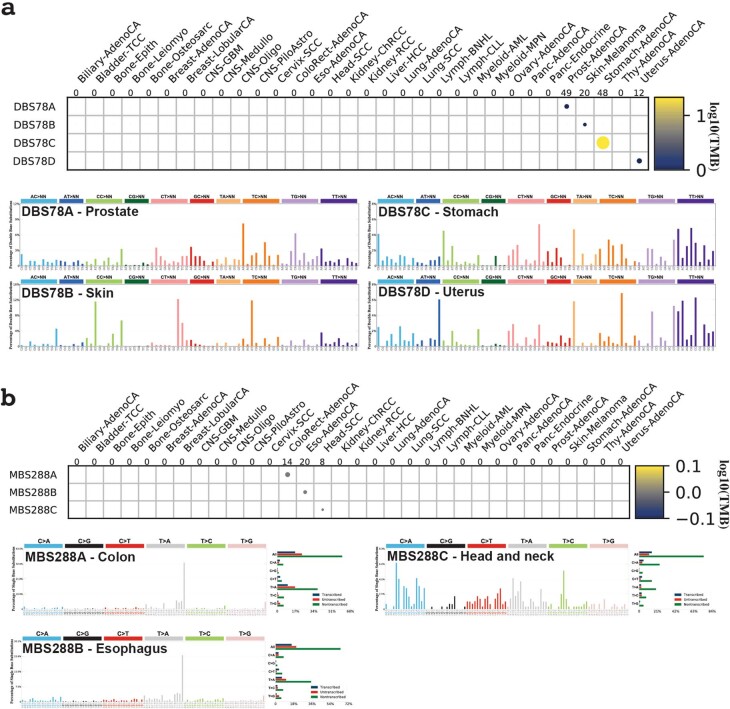
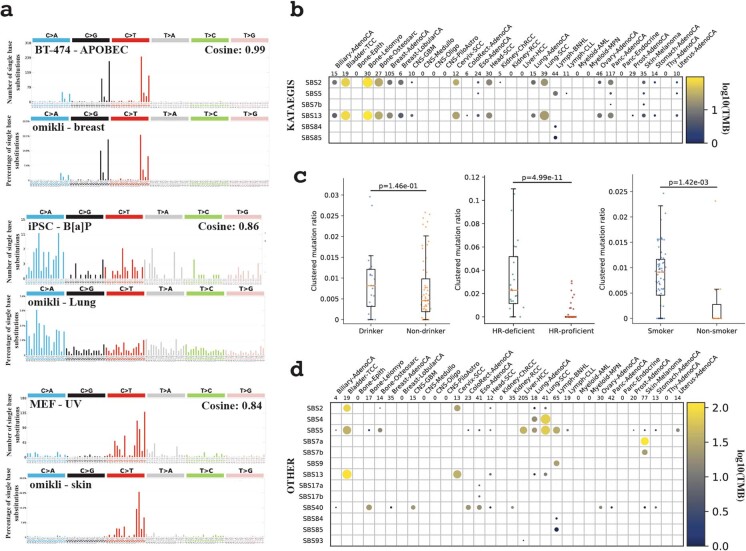
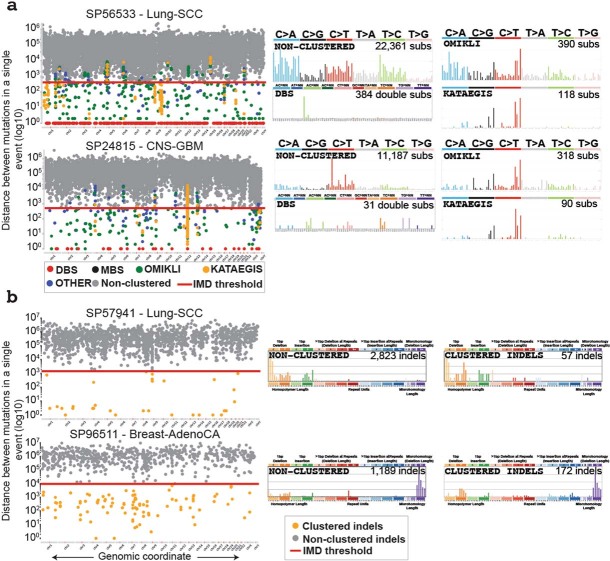
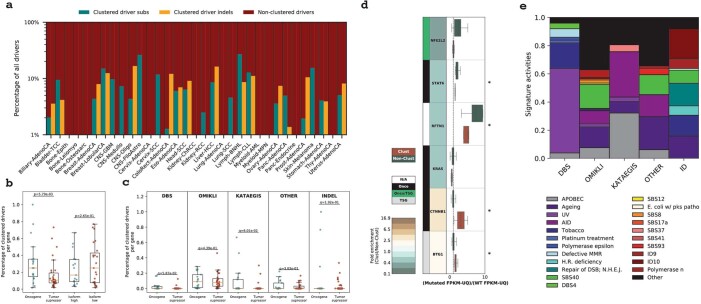
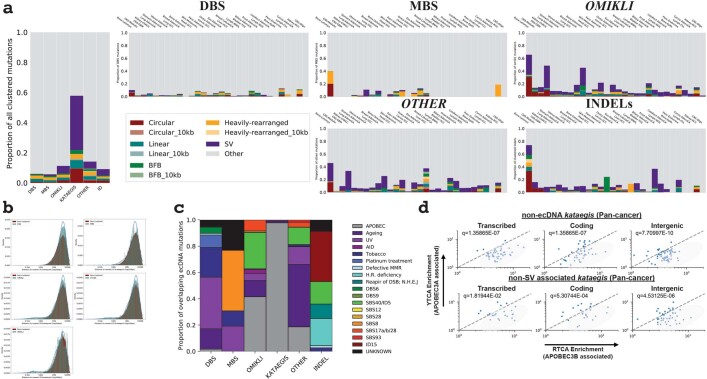
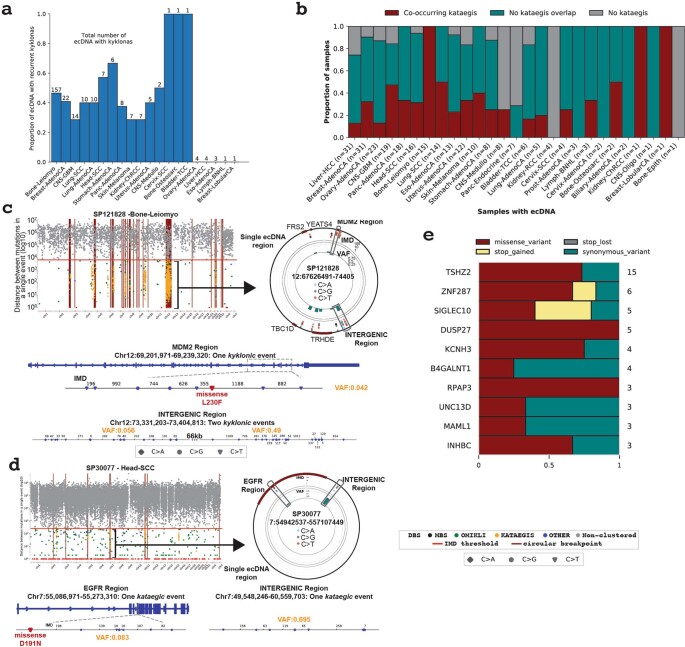
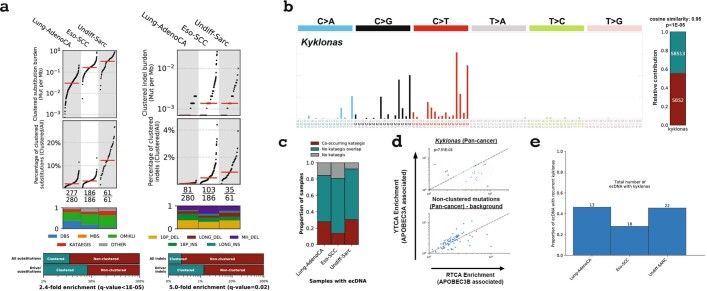
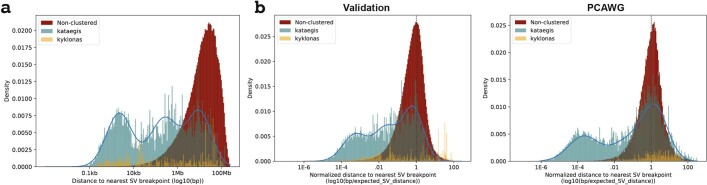
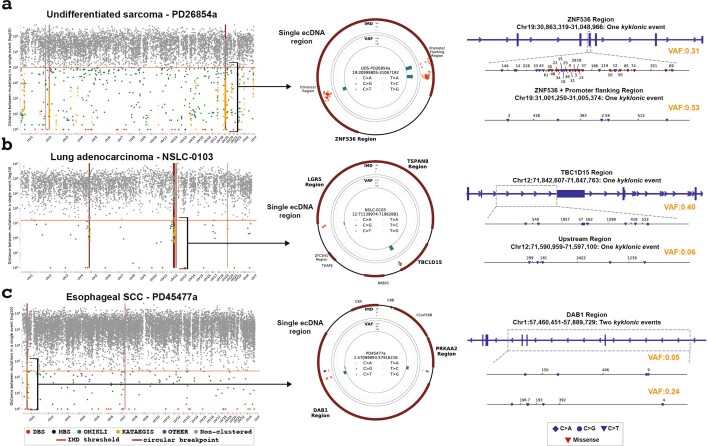
Clustered somatic mutations are common in cancer genomes and previous analyses reveal several types of clustered single-base substitutions, which include doublet- and multi-base substitutions1-5, diffuse hypermutation termed omikli6, and longer strand-coordinated events termed kataegis3,7-9. Here we provide a comprehensive characterization of clustered substitutions and clustered small insertions and deletions (indels) across 2,583 whole-genome-sequenced cancers from 30 types of cancer10. Clustered mutations were highly enriched in driver genes and associated with differential gene expression and changes in overall survival. Several distinct mutational processes gave rise to clustered indels, including signatures that were enriched in tobacco smokers and homologous-recombination-deficient cancers. Doublet-base substitutions were caused by at least 12 mutational processes, whereas most multi-base substitutions were generated by either tobacco smoking or exposure to ultraviolet light. Omikli events, which have previously been attributed to APOBEC3 activity6, accounted for a large proportion of clustered substitutions; however, only 16.2% of omikli matched APOBEC3 patterns. Kataegis was generated by multiple mutational processes, and 76.1% of all kataegic events exhibited mutational patterns that are associated with the activation-induced deaminase (AID) and APOBEC3 family of deaminases. Co-occurrence of APOBEC3 kataegis and extrachromosomal DNA (ecDNA), termed kyklonas (Greek for cyclone), was found in 31% of samples with ecDNA. Multiple distinct kyklonic events were observed on most mutated ecDNA. ecDNA containing known cancer genes exhibited both positive selection and kyklonic hypermutation. Our results reveal the diversity of clustered mutational processes in human cancer and the role of APOBEC3 in recurrently mutating and fuelling the evolution of ecDNA.
© 2022. The Author(s).
Conflict of interest statement
M.P. is a shareholder in Vertex Pharmaceuticals. V.B. is a co-founder, consultant and Scientific Advisory Board member of, and has equity interest in, Boundless Bio, and Abterra. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict-of-interest policies. E.N.B. and L.B.A. declare filing a provisional patent application for using clustered mutations as clinical prognostic biomarkers in cancer. P.S.M. is a co-founder of Boundless Bio. He has equity in the company and he chairs the Scientific Advisory Board, for which he is compensated. L.B.A. is an inventor on US patent no. 10,776,718 for source identification by non-negative matrix factorization. All other authors declare no competing interests.
Figures
References
-
- de Gruijl FR, van Kranen HJ, Mullenders LH. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol. B. 2001;63:19–27. - PubMed
