

DECIPHER: Supporting the interpretation and sharing of rare disease phenotype-linked variant data to advance diagnosis and research
- PMID: 35143074
- PMCID: PMC9303633
- DOI: 10.1002/humu.24340
DECIPHER: Supporting the interpretation and sharing of rare disease phenotype-linked variant data to advance diagnosis and research
Abstract
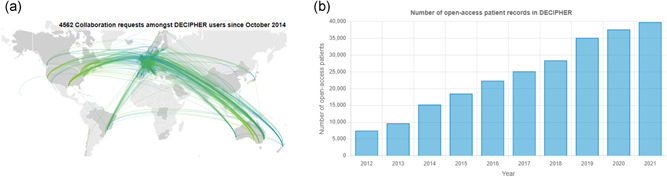
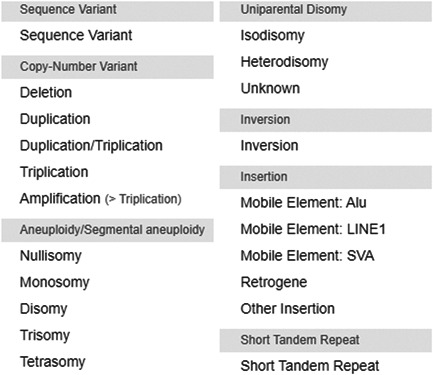
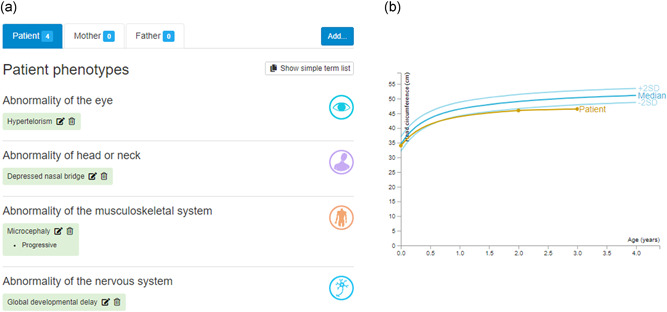
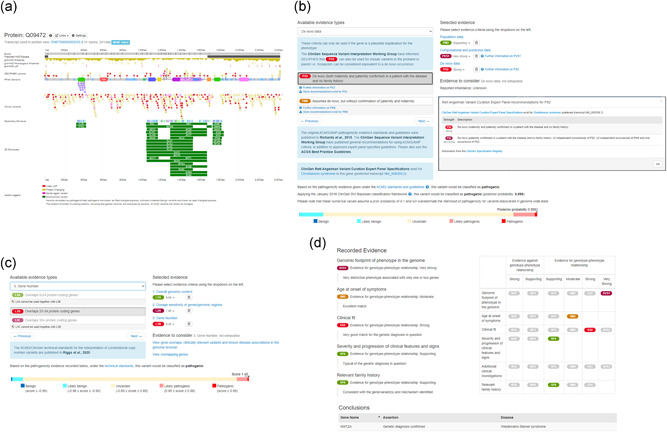
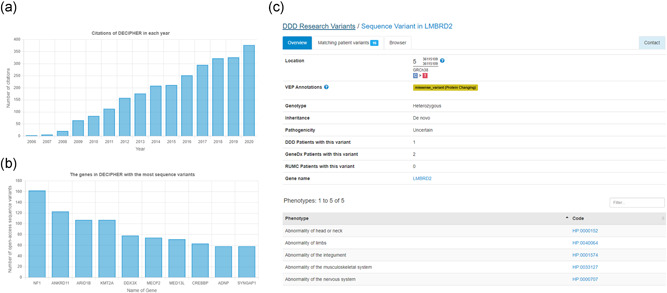
DECIPHER (https://www.deciphergenomics.org) is a free web platform for sharing anonymized phenotype-linked variant data from rare disease patients. Its dynamic interpretation interfaces contextualize genomic and phenotypic data to enable more informed variant interpretation, incorporating international standards for variant classification. DECIPHER supports almost all types of germline and mosaic variation in the nuclear and mitochondrial genome: sequence variants, short tandem repeats, copy-number variants, and large structural variants. Patient phenotypes are deposited using Human Phenotype Ontology (HPO) terms, supplemented by quantitative data, which is aggregated to derive gene-specific phenotypic summaries. It hosts data from >250 projects from ~40 countries, openly sharing >40,000 patient records containing >51,000 variants and >172,000 phenotype terms. The rich phenotype-linked variant data in DECIPHER drives rare disease research and diagnosis by enabling patient matching within DECIPHER and with other resources, and has been cited in >2,600 publications. In this study, we describe the types of data deposited to DECIPHER, the variant interpretation tools, and patient matching interfaces which make DECIPHER an invaluable rare disease resource.
Keywords: Matchmaker Exchange; genetic disorders; genomic medicine; genotype phenotype correlation; rare diseases; variant interpretation; whole-exome sequencing; whole-genome sequencing.
© 2022 The Authors. Human Mutation published by Wiley Periodicals LLC.
Conflict of interest statement
Matthew Hurles is a cofounder, shareholder, and nonexecutive director of Congenica Ltd., a diagnostic software company.
Figures


References
-
- Alsharhan, H. , He, M. , Edmondson, A. C. , Daniel, E. , Chen, J. , Donald, T. , Bakhtiari, S. , Amor, D. J. , Jones, E. A. , Vassallo, G. , Vincent, M. , Cogné, B. , Deb, W. , Werners, A. H. , Jin, S. C. , Bilguvar, K. , Christodoulou, J. , Webster, R. I. , Yearwood, K. R. , … Sobering, A. K. (2021). ALG13 X‐linked intellectual disability: New variants, glycosylation analysis, and expanded phenotypes. Journal of Inherited Metabolic Disease, 44(4), 1001–1012. 10.1002/jimd.12378 - DOI - PMC - PubMed
-
- Arachchi, H. , Wojcik, M. H. , Weisburd, B. , Jacobsen, J. , Valkanas, E. , Baxter, S. , Byrne, A. B. , O'Donnell‐Luria, A. H. , Haendel, M. , Smedley, D. , MacArthur, D. G. , Philippakis, A. A. , & Rehm, H. L. (2018). matchbox: An open‐source tool for patient matching via the Matchmaker Exchange. Human Mutation, 39(12), 1827–1834. 10.1002/humu.23655 - DOI - PMC - PubMed
-
- Balasubramanian, M. , Dingemans, A. , Albaba, S. , Richardson, R. , Yates, T. M. , Cox, H. , Douzgou, S. , Armstrong, R. , Sansbury, F. H. , Burke, K. B. , Fry, A. E. , Ragge, N. , Sharif, S. , Foster, A. , De Sandre‐Giovannoli, A. , Elouej, S. , Vasudevan, P. , Mansour, S. , Wilson, K. , … Kleefstra, T. (2021). Comprehensive study of 28 individuals with SIN3A‐related disorder underscoring the associated mild cognitive and distinctive facial phenotype. European Journal of Human Genetics, 29(4), 625–636. 10.1038/s41431-020-00769-7 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
