

A mammalian methylation array for profiling methylation levels at conserved sequences
- PMID: 35145108
- PMCID: PMC8831611
- DOI: 10.1038/s41467-022-28355-z
A mammalian methylation array for profiling methylation levels at conserved sequences
Abstract
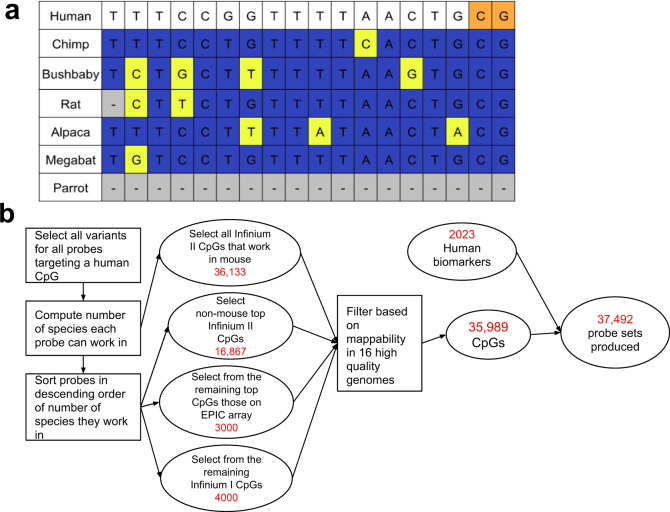
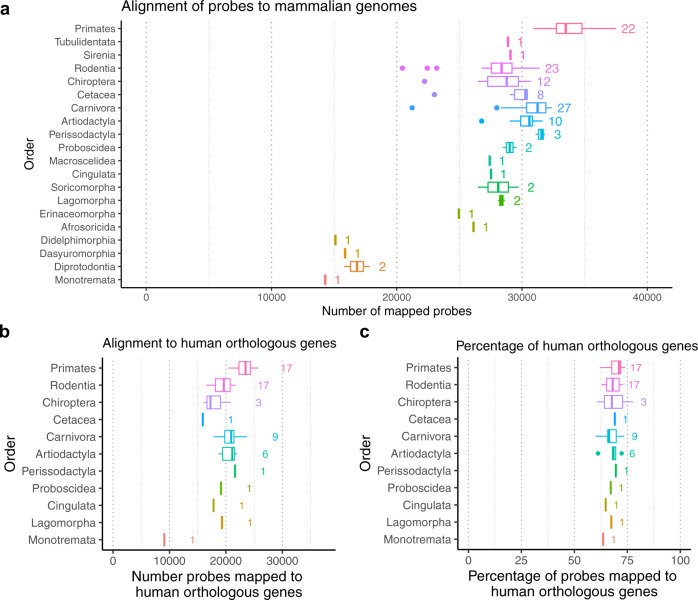
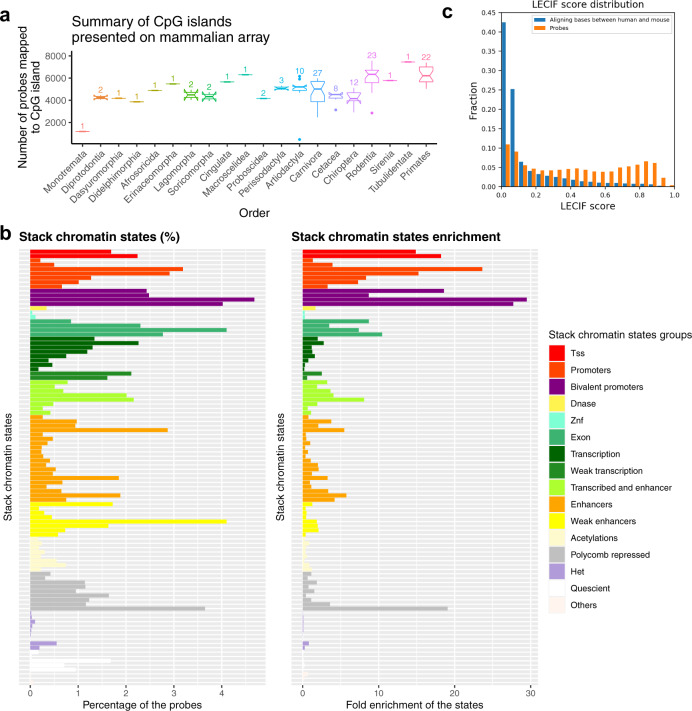
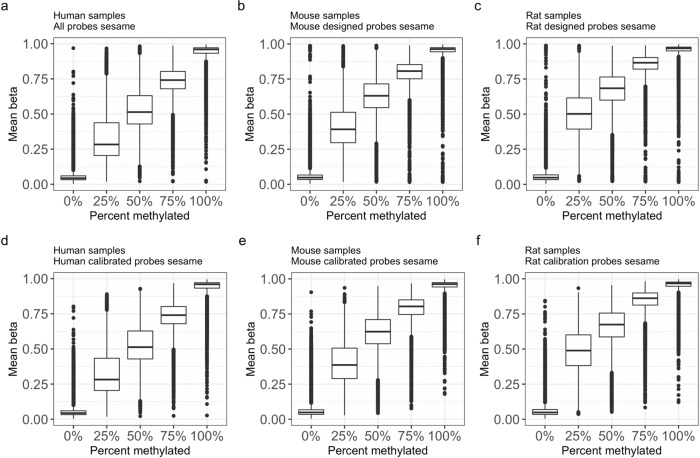
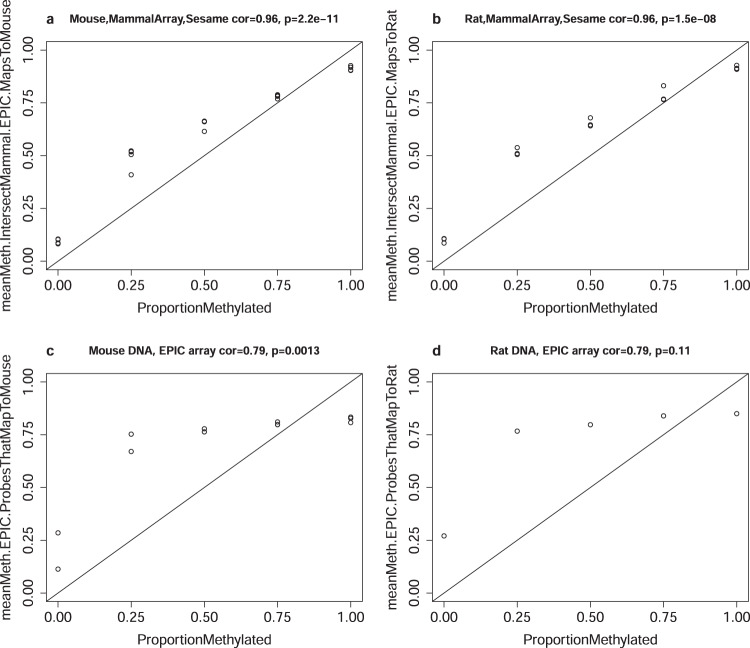
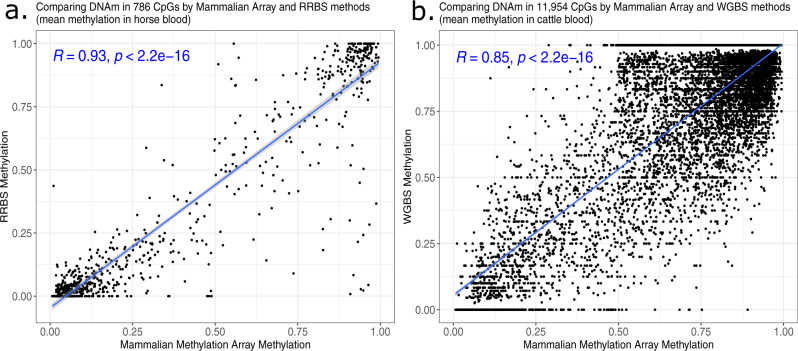
Infinium methylation arrays are not available for the vast majority of non-human mammals. Moreover, even if species-specific arrays were available, probe differences between them would confound cross-species comparisons. To address these challenges, we developed the mammalian methylation array, a single custom array that measures up to 36k CpGs per species that are well conserved across many mammalian species. We designed a set of probes that can tolerate specific cross-species mutations. We annotate the array in over 200 species and report CpG island status and chromatin states in select species. Calibration experiments demonstrate the high fidelity in humans, rats, and mice. The mammalian methylation array has several strengths: it applies to all mammalian species even those that have not yet been sequenced, it provides deep coverage of conserved cytosines facilitating the development of epigenetic biomarkers, and it increases the probability that biological insights gained in one species will translate to others.
© 2022. The Author(s).
Conflict of interest statement
The Regents of the University of California filed a patent application (publication number WO2020150705) related to this work for which A.A., B.B., J.E. and S.H. are named inventors. S.H. is a founder of the non-profit Epigenetic Clock Development Foundation, which has licensed several patents from his employer UC Regents, and distributes the mammalian methylation array. Bret Barnes is an employee for Illumina Inc which manufactures the mammalian methylation array. The remaining authors declare no competing interests.
Figures
References
-
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat. Rev. Genet. 2013;14:204–220. - PubMed
-
- Bibikova, M. et al. Genome-wide DNA methylation profiling using Infinium((R)) assay. Epigenomics. 1, 10.2217/epi.09.14 (2009). - PubMed
-
- Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
