

METTL16 exerts an m6A-independent function to facilitate translation and tumorigenesis
- PMID: 35145225
- PMCID: PMC9070413
- DOI: 10.1038/s41556-021-00835-2
METTL16 exerts an m6A-independent function to facilitate translation and tumorigenesis
Abstract
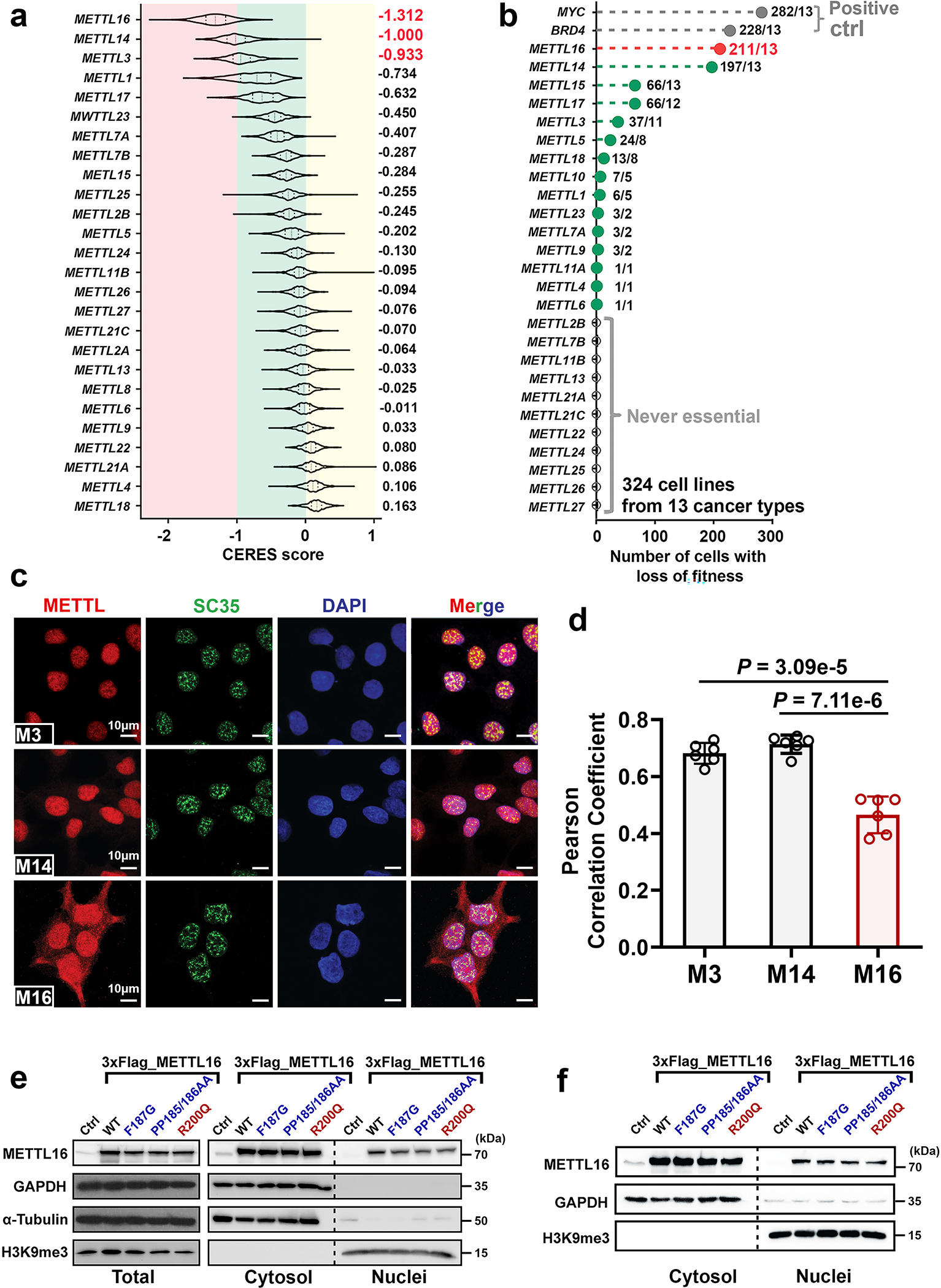
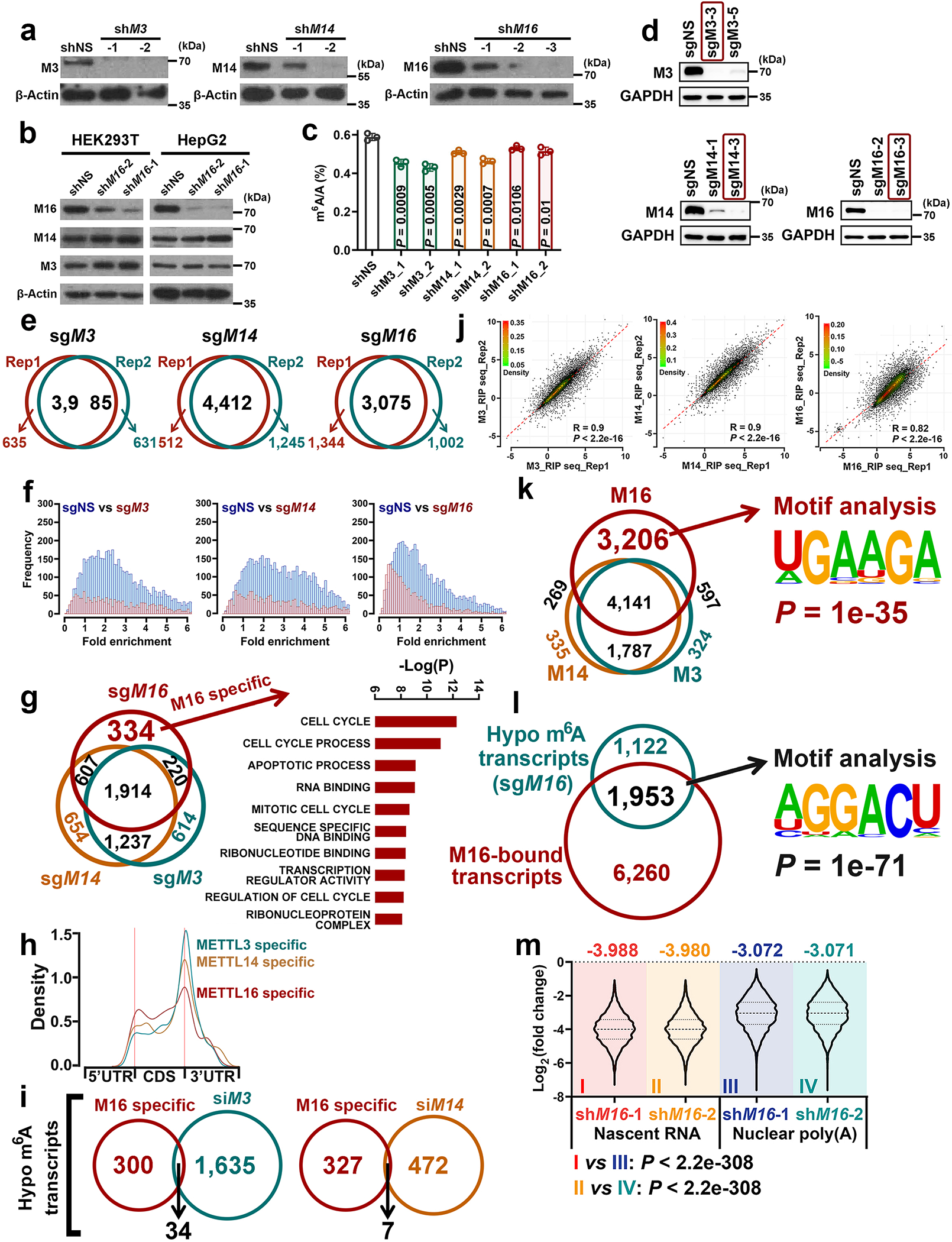
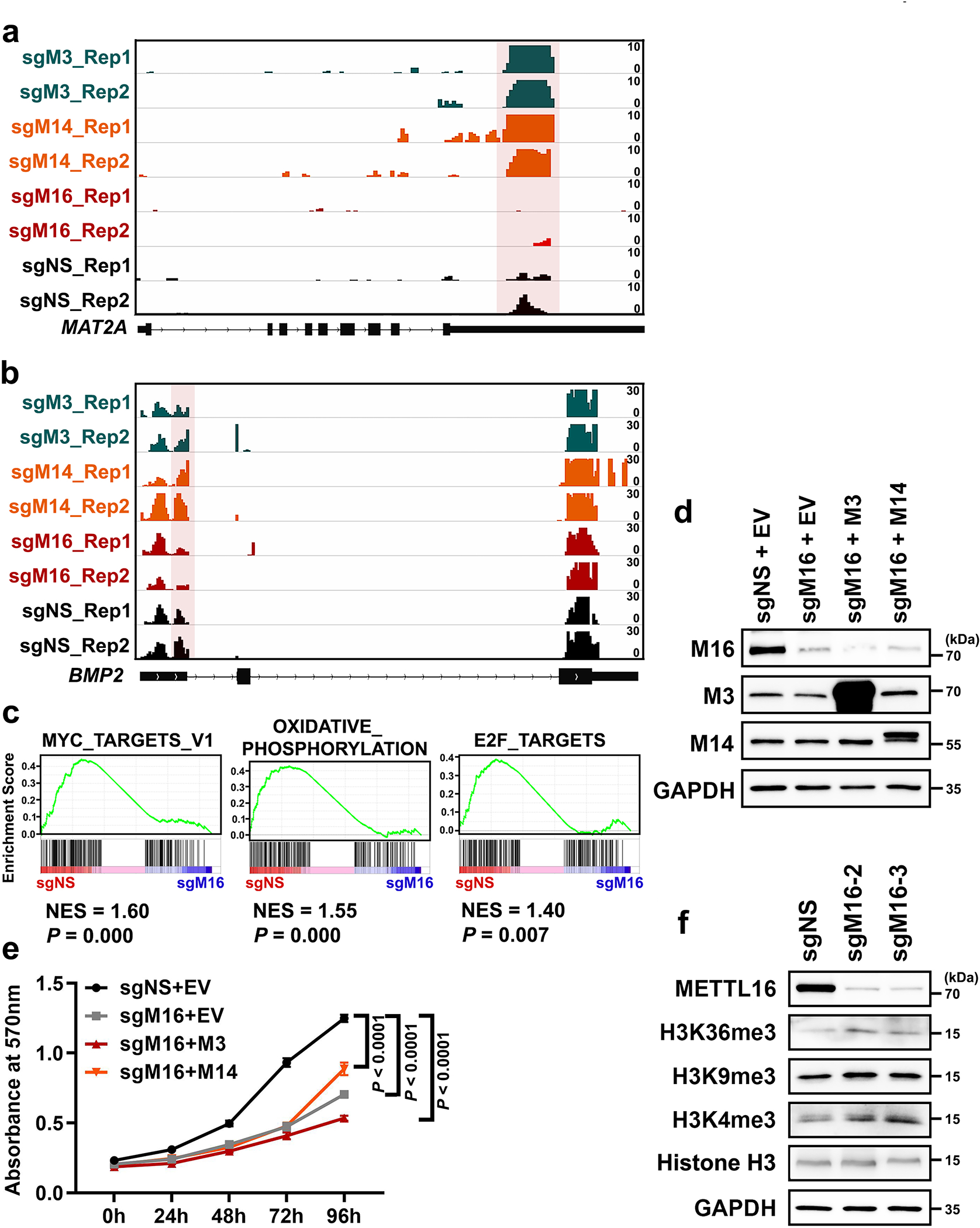
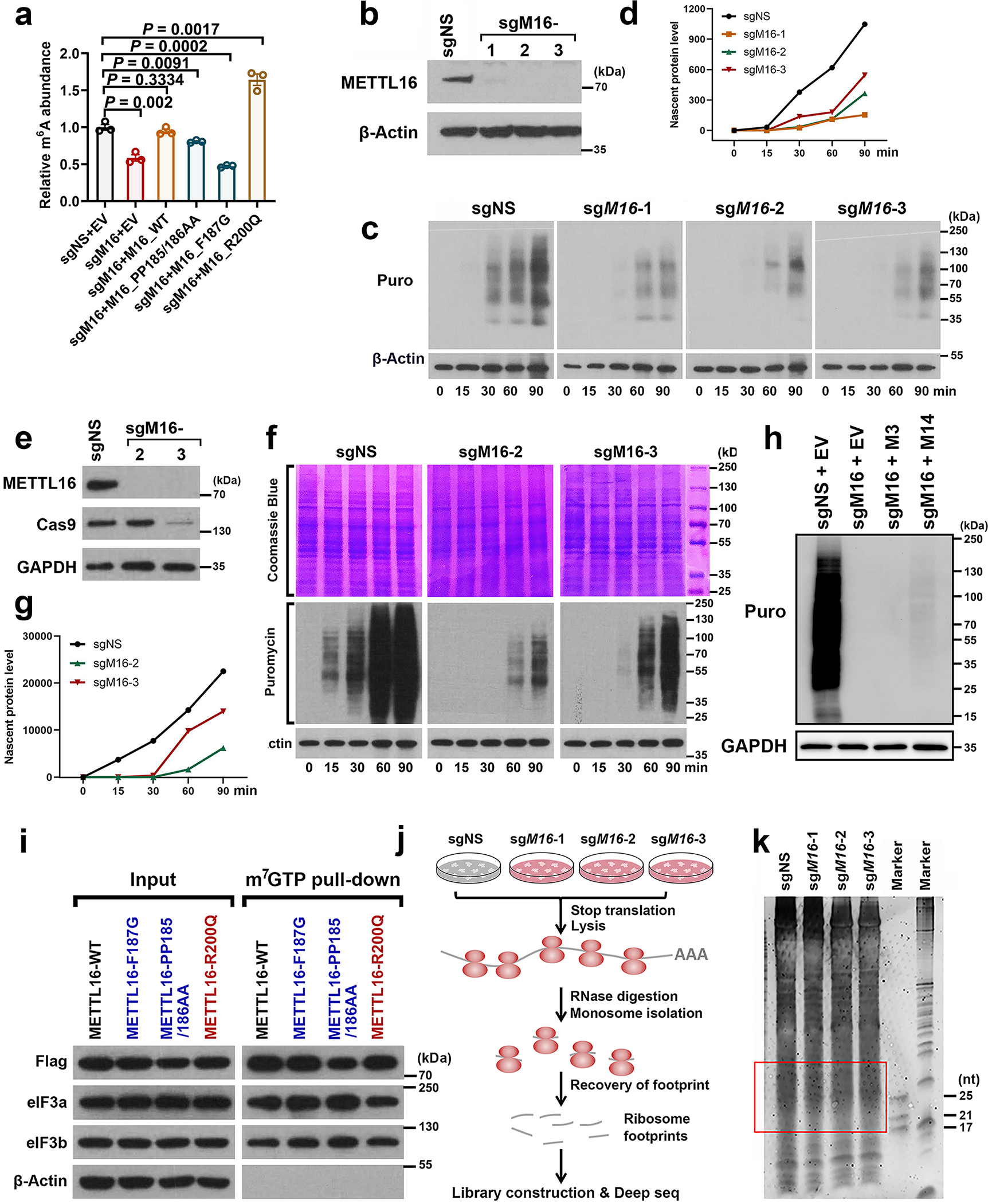
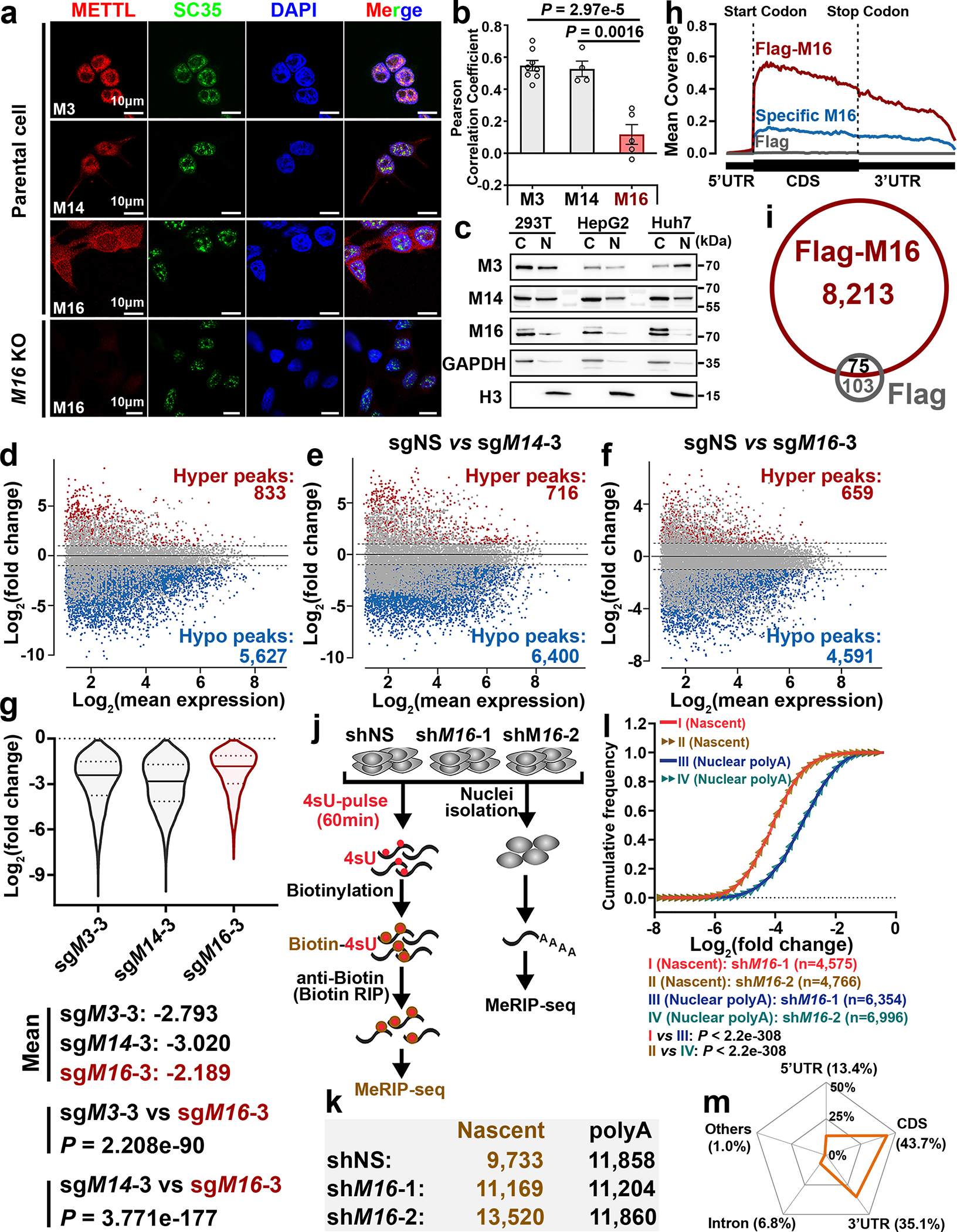
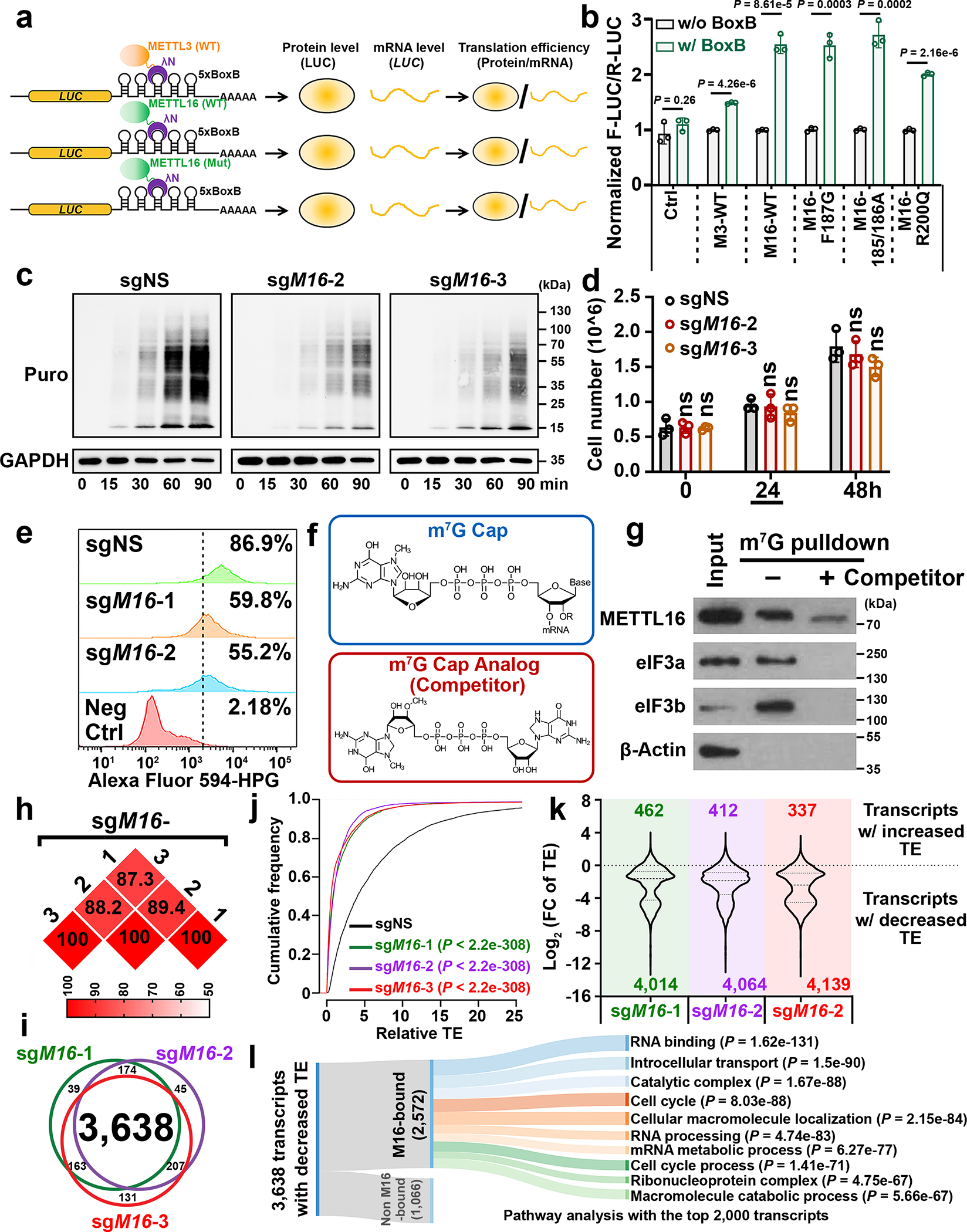
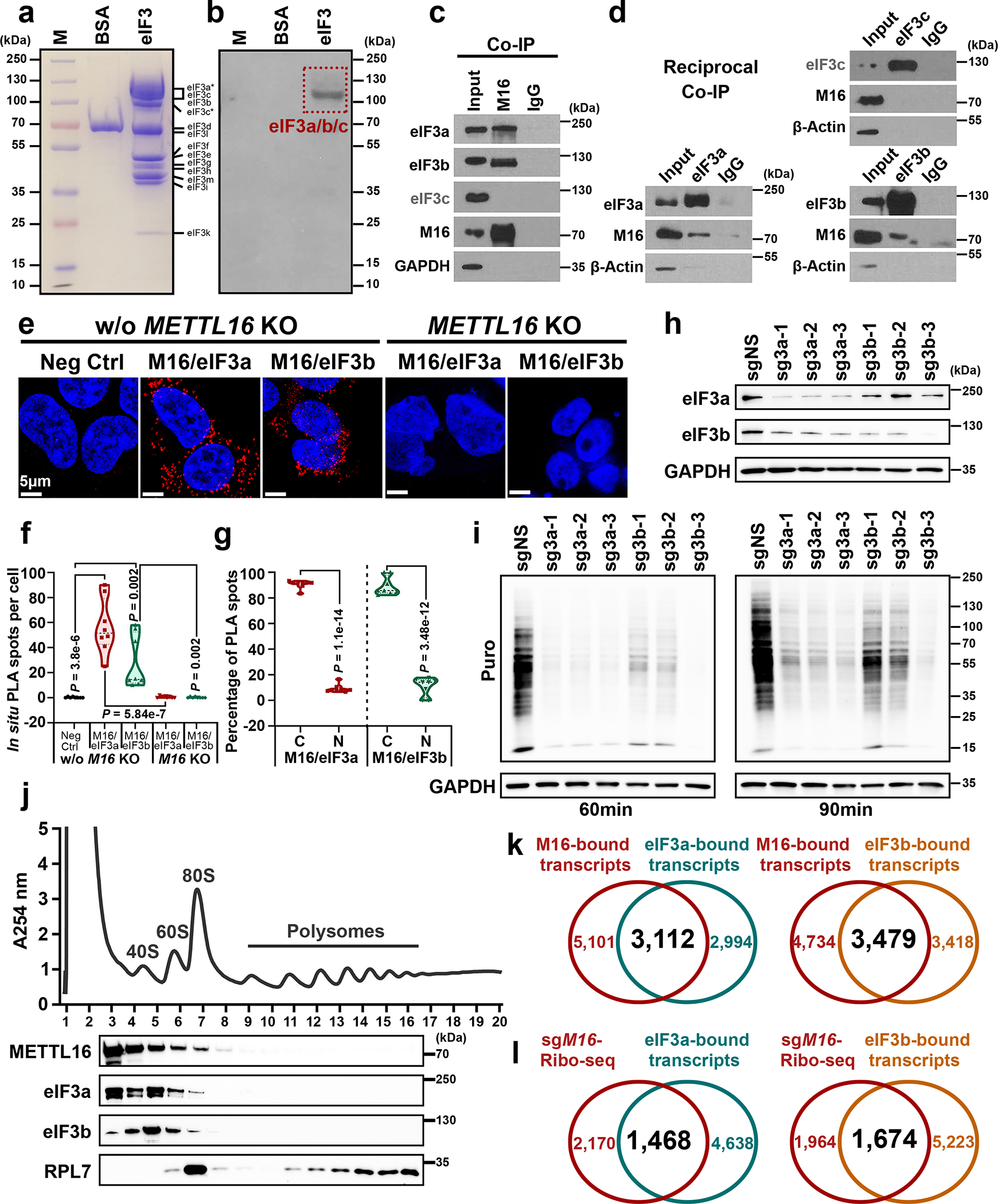
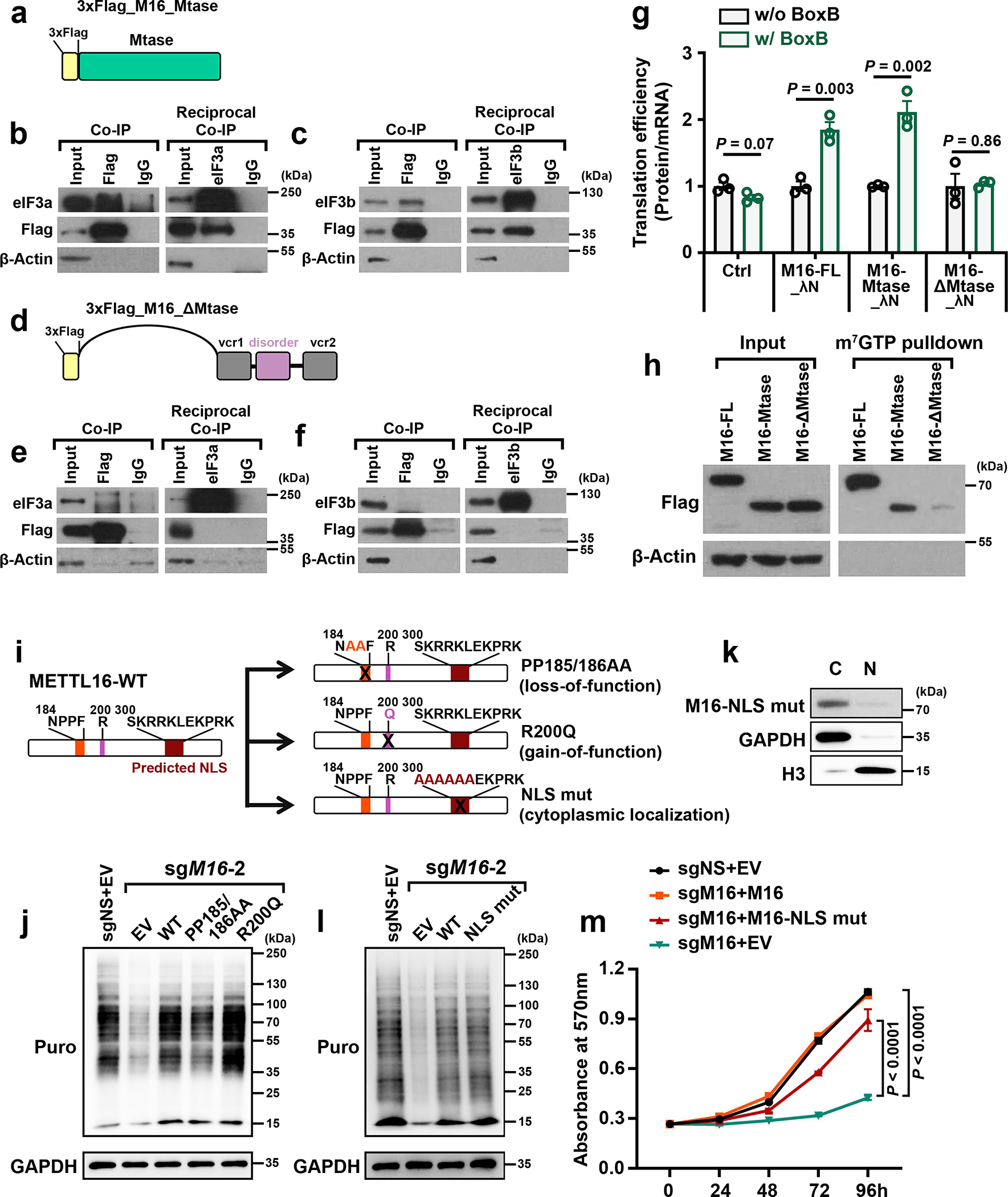
METTL16 has recently been identified as an RNA methyltransferase responsible for the deposition of N6-methyladenosine (m6A) in a few transcripts. Whether METTL16 methylates a large set of transcripts, similar to METTL3 and METTL14, remains unclear. Here we show that METTL16 exerts both methyltransferase activity-dependent and -independent functions in gene regulation. In the cell nucleus, METTL16 functions as an m6A writer to deposit m6A into hundreds of its specific messenger RNA targets. In the cytosol, METTL16 promotes translation in an m6A-independent manner. More specifically, METTL16 directly interacts with the eukaryotic initiation factors 3a and -b as well as ribosomal RNA through its Mtase domain, thereby facilitating the assembly of the translation-initiation complex and promoting the translation of over 4,000 mRNA transcripts. Moreover, we demonstrate that METTL16 is critical for the tumorigenesis of hepatocellular carcinoma. Collectively, our studies reveal previously unappreciated dual functions of METTL16 as an m6A writer and a translation-initiation facilitator, which together contribute to its essential function in tumorigenesis.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
C.H. is a scientific founder and a scientific advisory board member of Accent Therapeutics, Inc., and holds equities with the company. J.C. is a scientific advisory board member of Race Oncology. The remaining authors declare no competing interests.
Figures
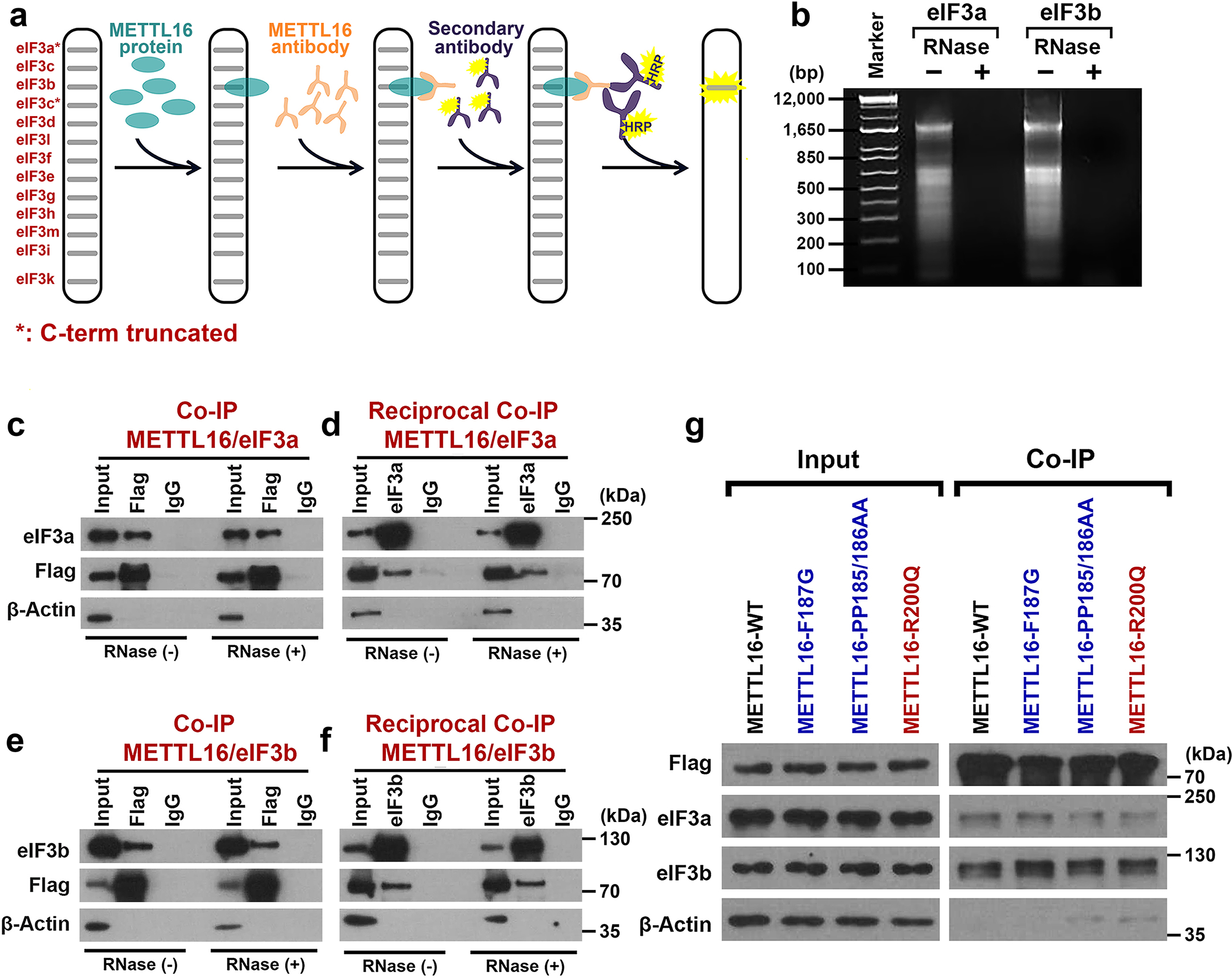
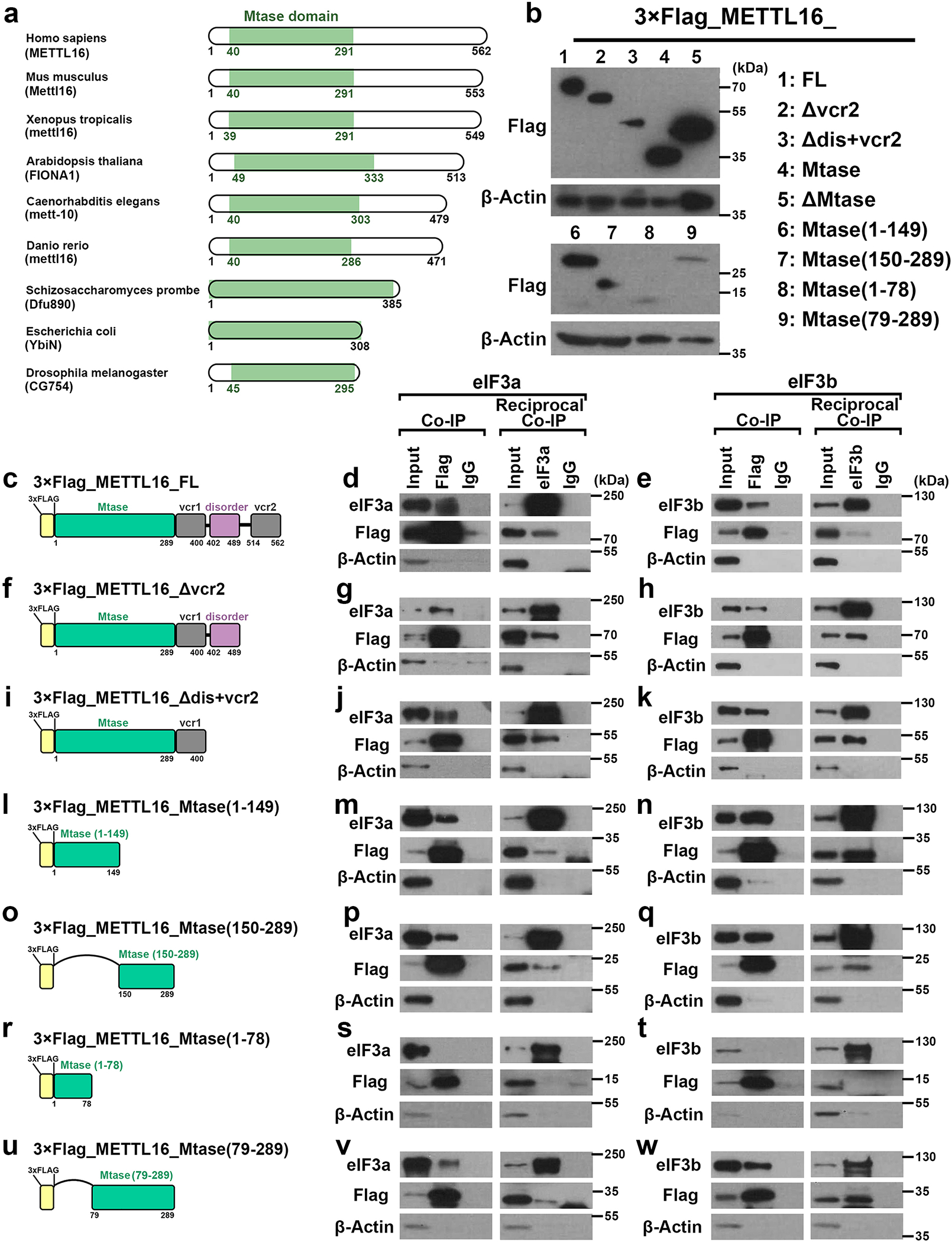
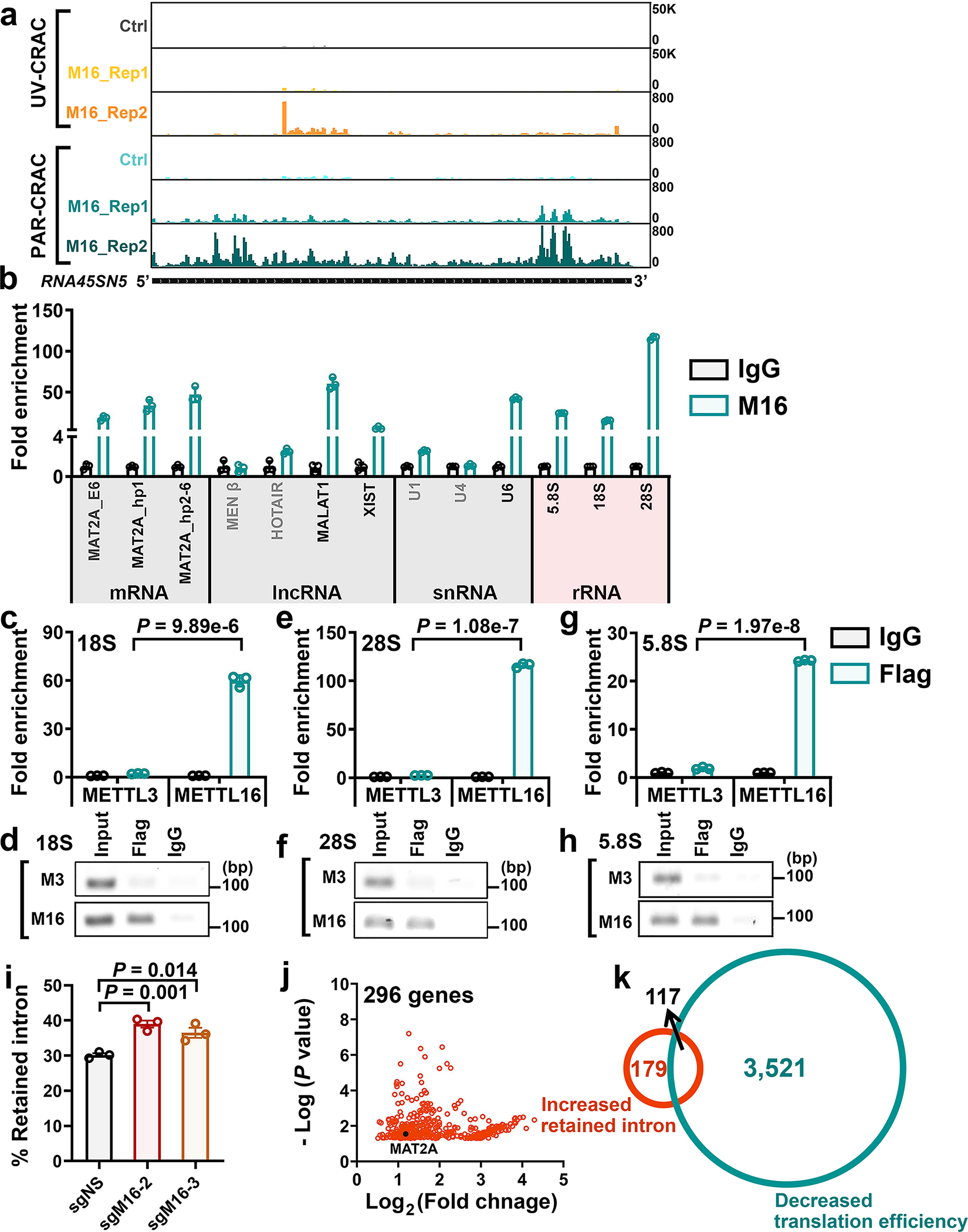
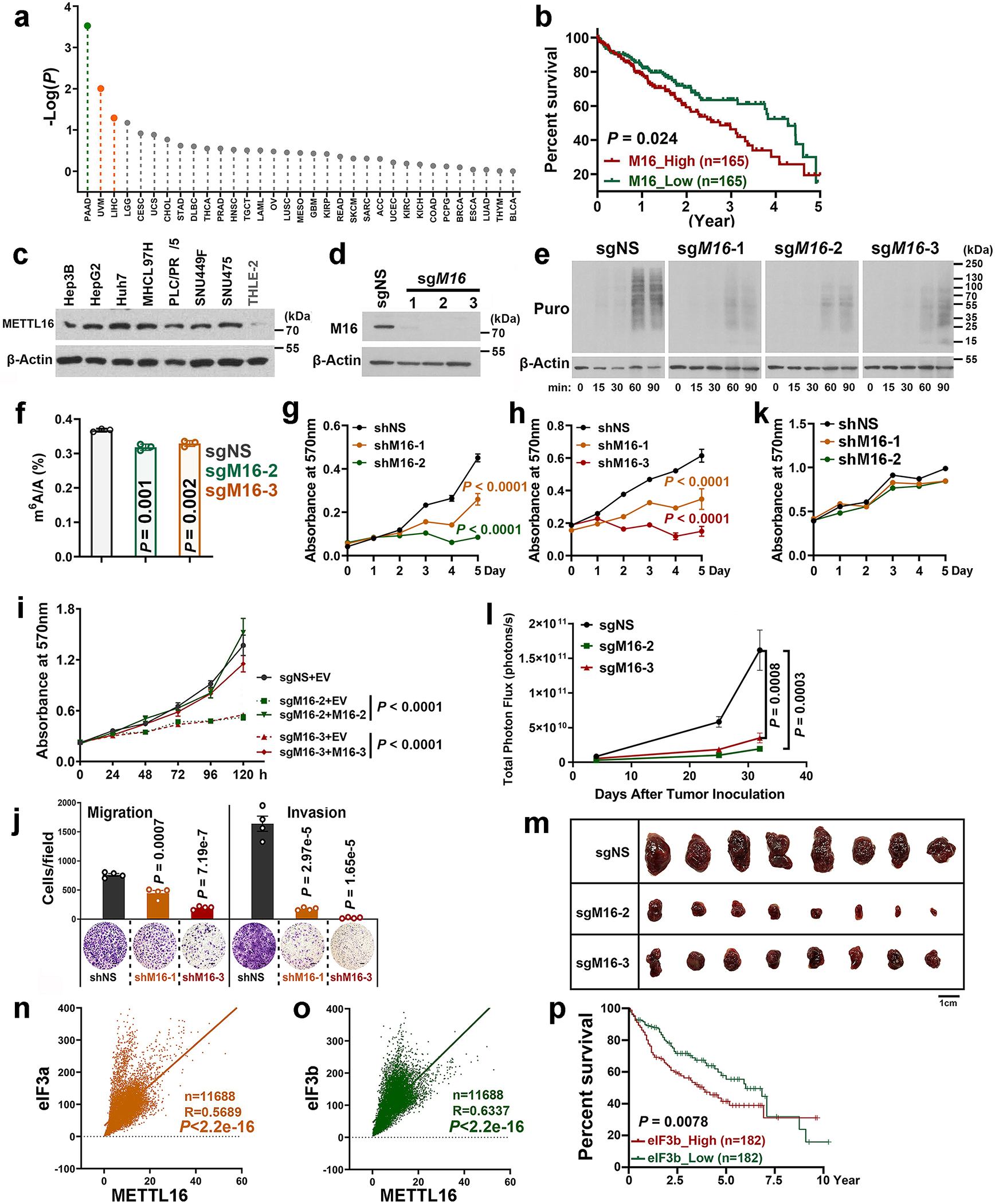
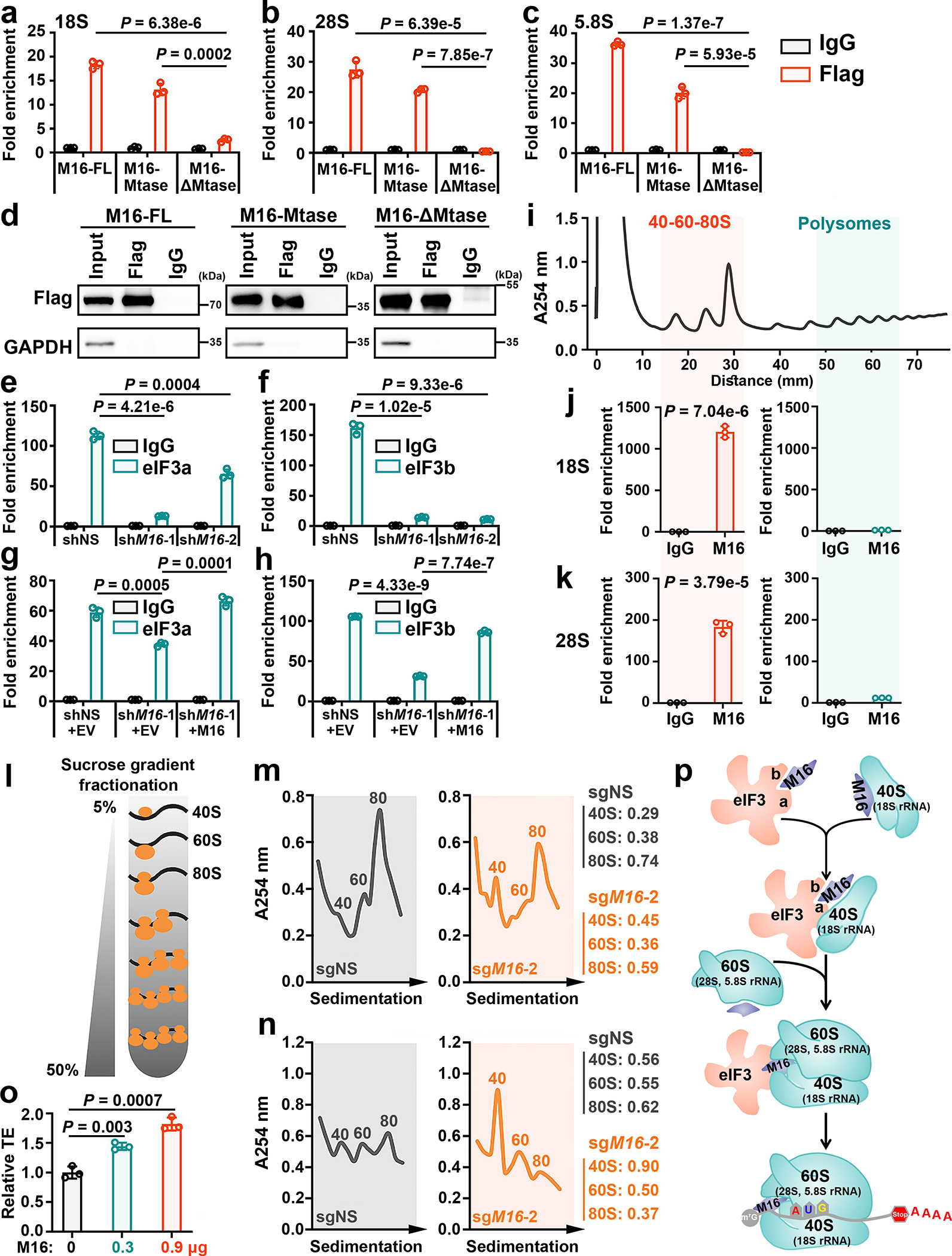
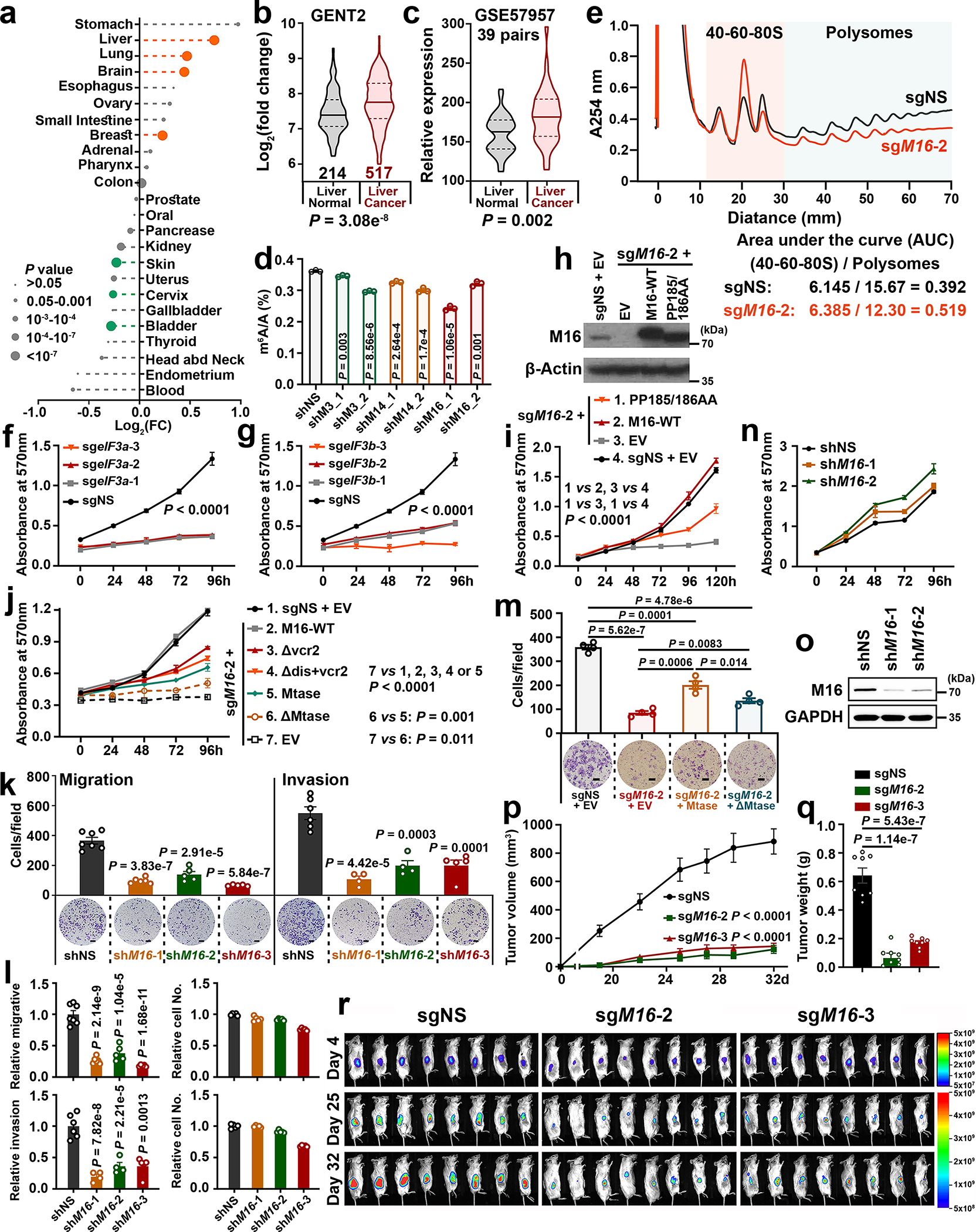
References
-
- Wang X et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- T32 HD007009/HD/NICHD NIH HHS/United States
- R01 CA236399/CA/NCI NIH HHS/United States
- R01 CA139158/CA/NCI NIH HHS/United States
- R01 CA214965/CA/NCI NIH HHS/United States
- R01 CA211614/CA/NCI NIH HHS/United States
- R56 DK120282/DK/NIDDK NIH HHS/United States
- R01 GM113194/GM/NIGMS NIH HHS/United States
- R01 CA243386/CA/NCI NIH HHS/United States
- RM1 HG008935/HG/NHGRI NIH HHS/United States
- R01 DK124627/DK/NIDDK NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32 CA186895/CA/NCI NIH HHS/United States
- R01 CA271497/CA/NCI NIH HHS/United States
- R01 GM088599/GM/NIGMS NIH HHS/United States
- R01 DK124116/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
