

Barcoded bulk QTL mapping reveals highly polygenic and epistatic architecture of complex traits in yeast
- PMID: 35147078
- PMCID: PMC8979589
- DOI: 10.7554/eLife.73983
Barcoded bulk QTL mapping reveals highly polygenic and epistatic architecture of complex traits in yeast
Abstract
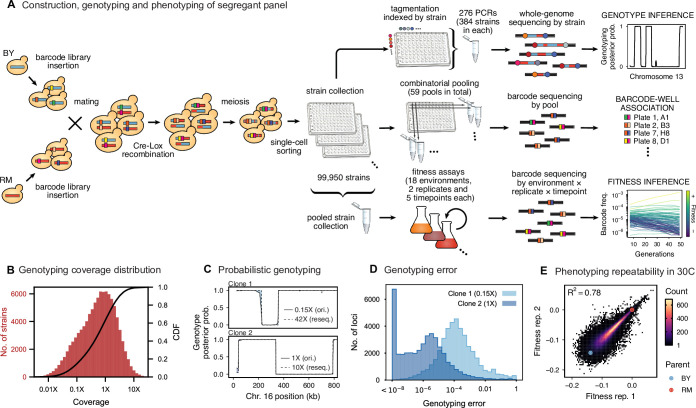
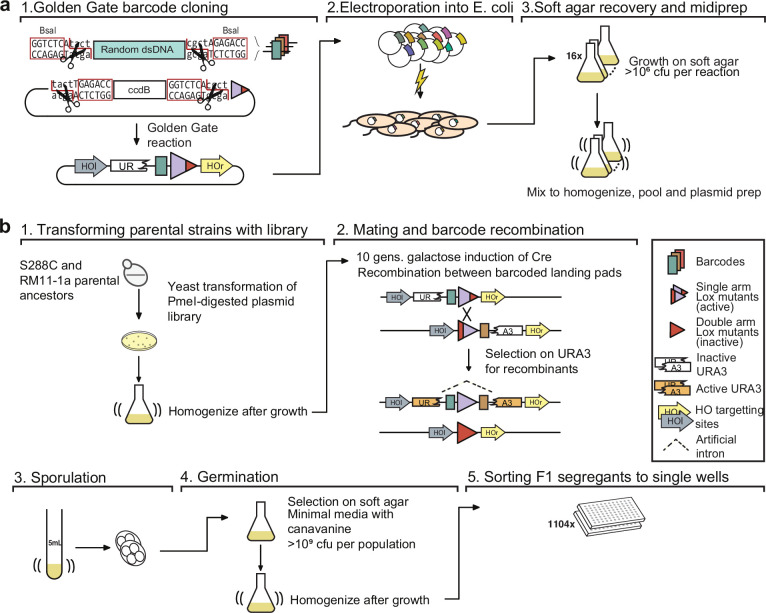
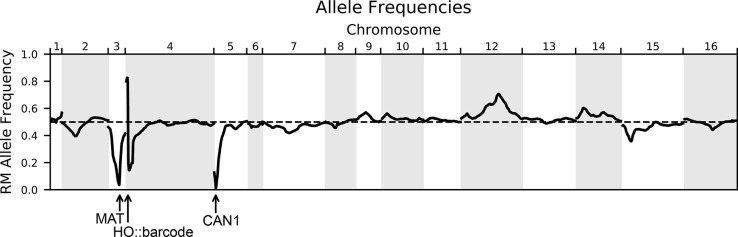
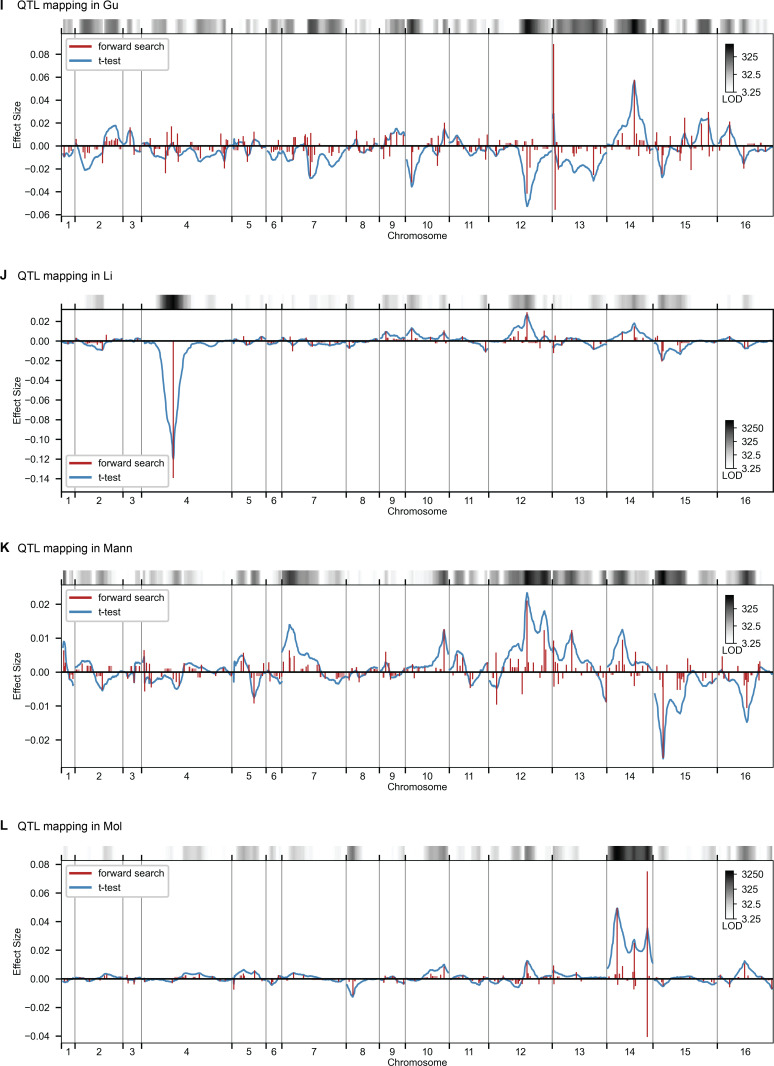
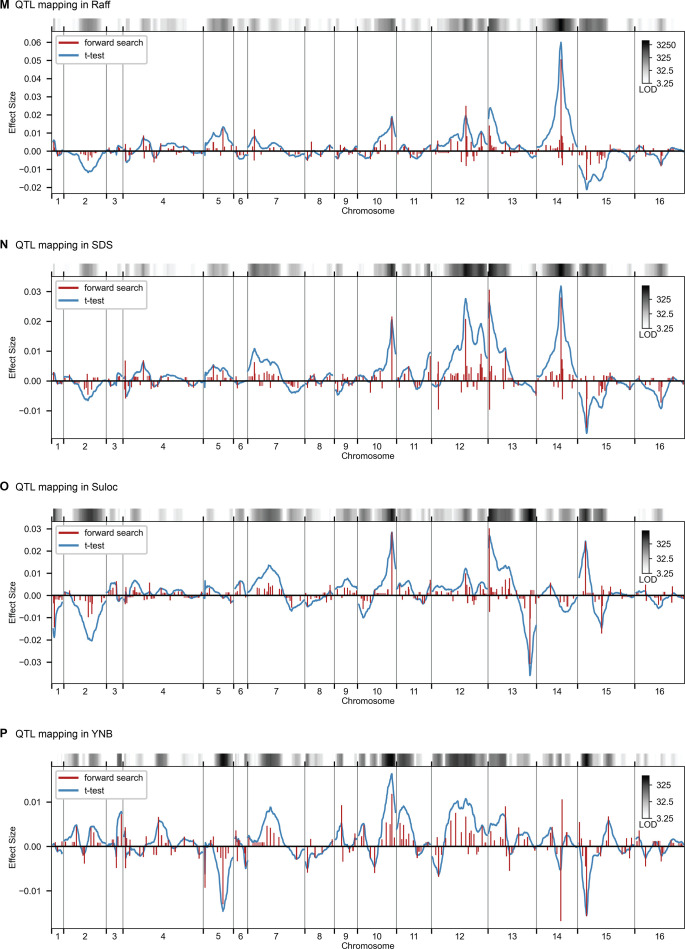
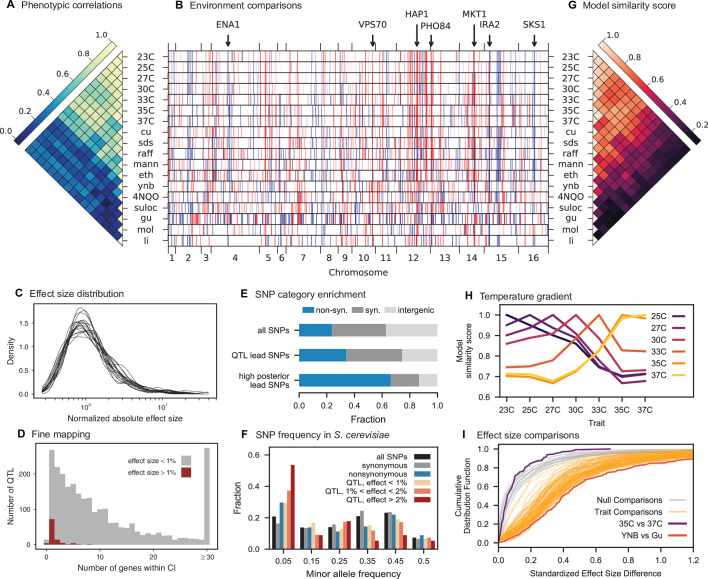
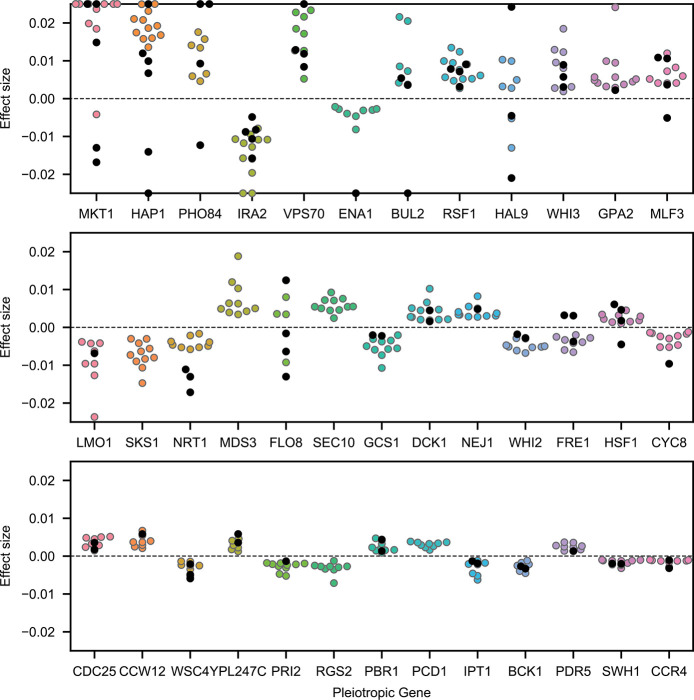
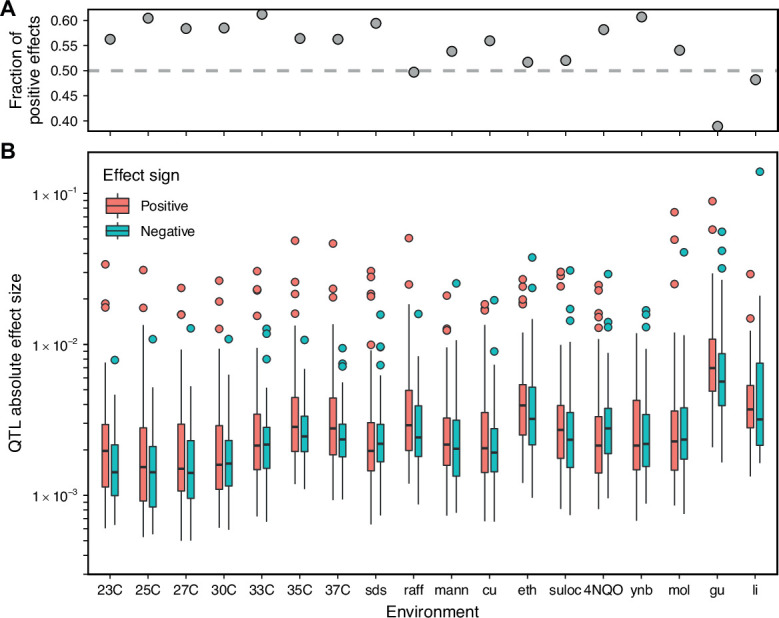
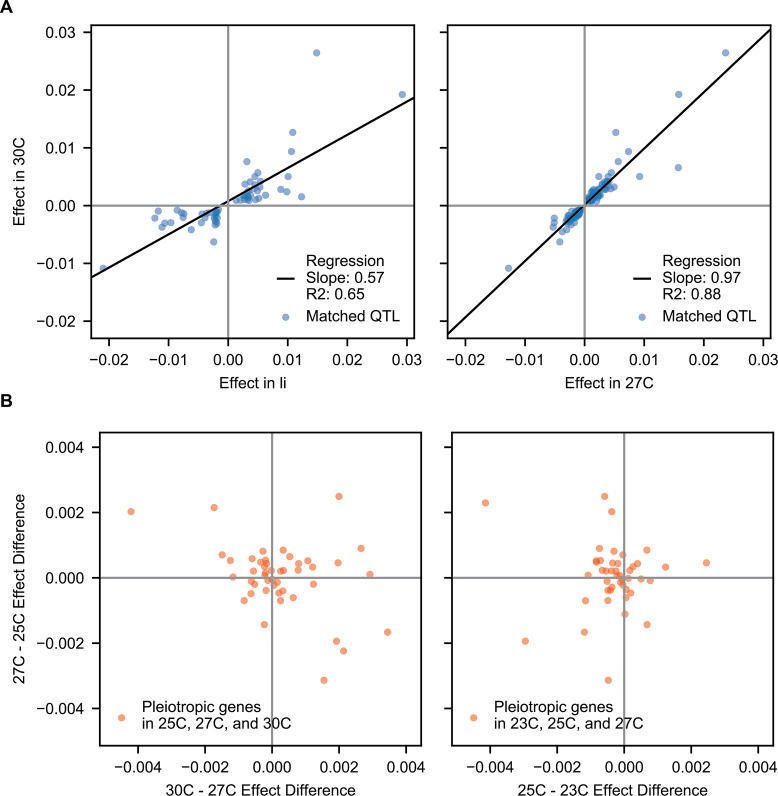
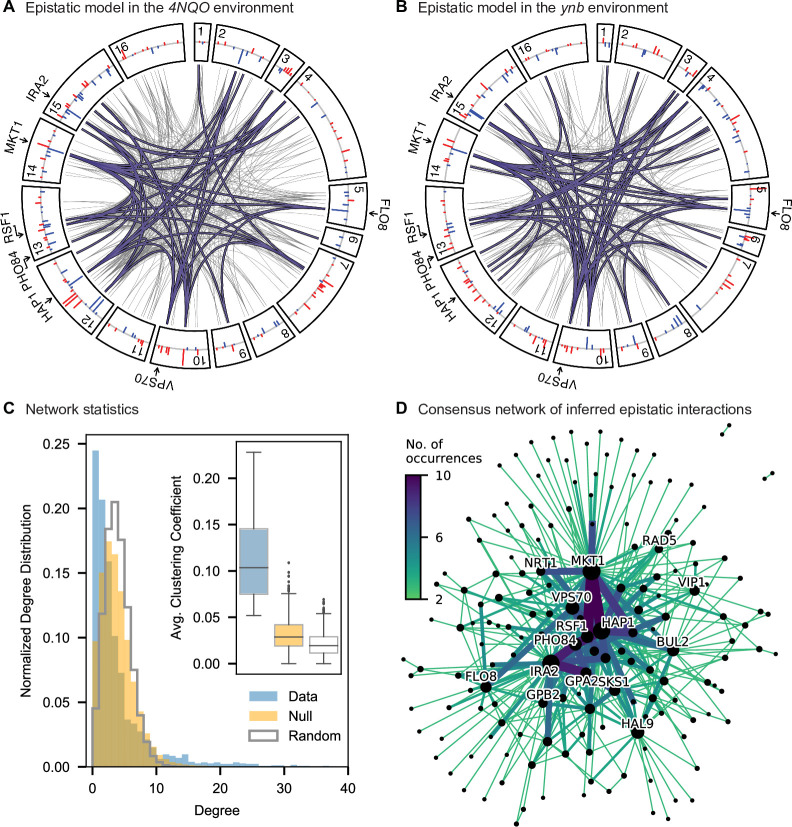
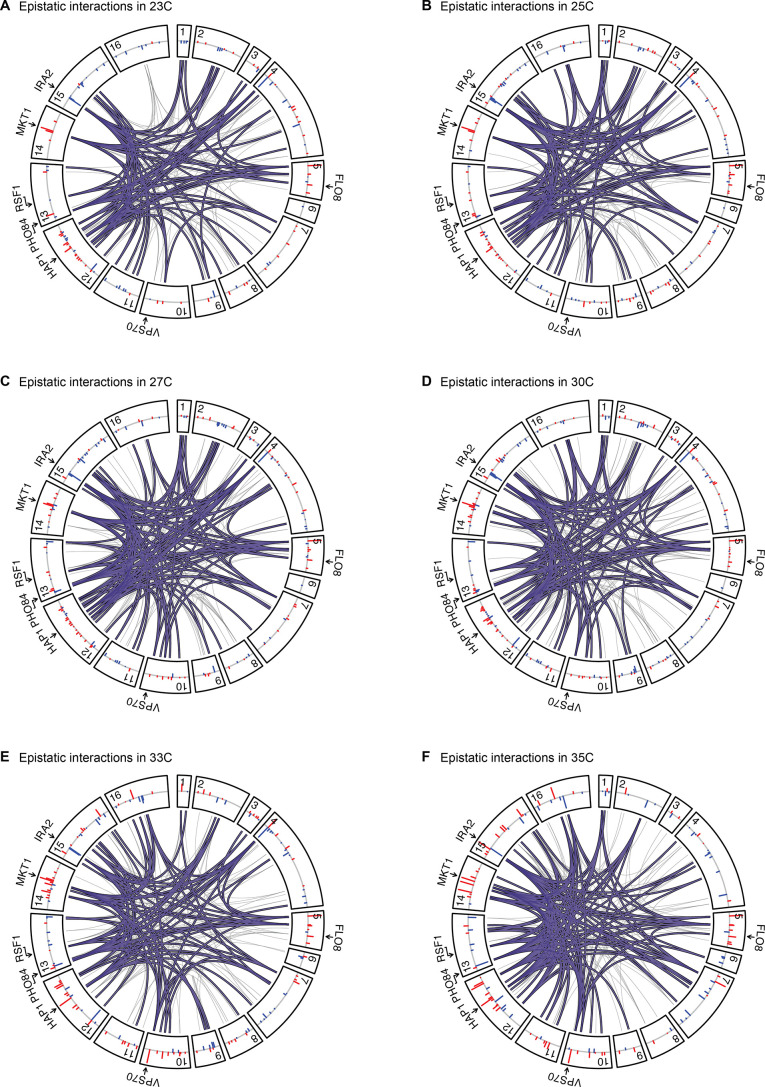
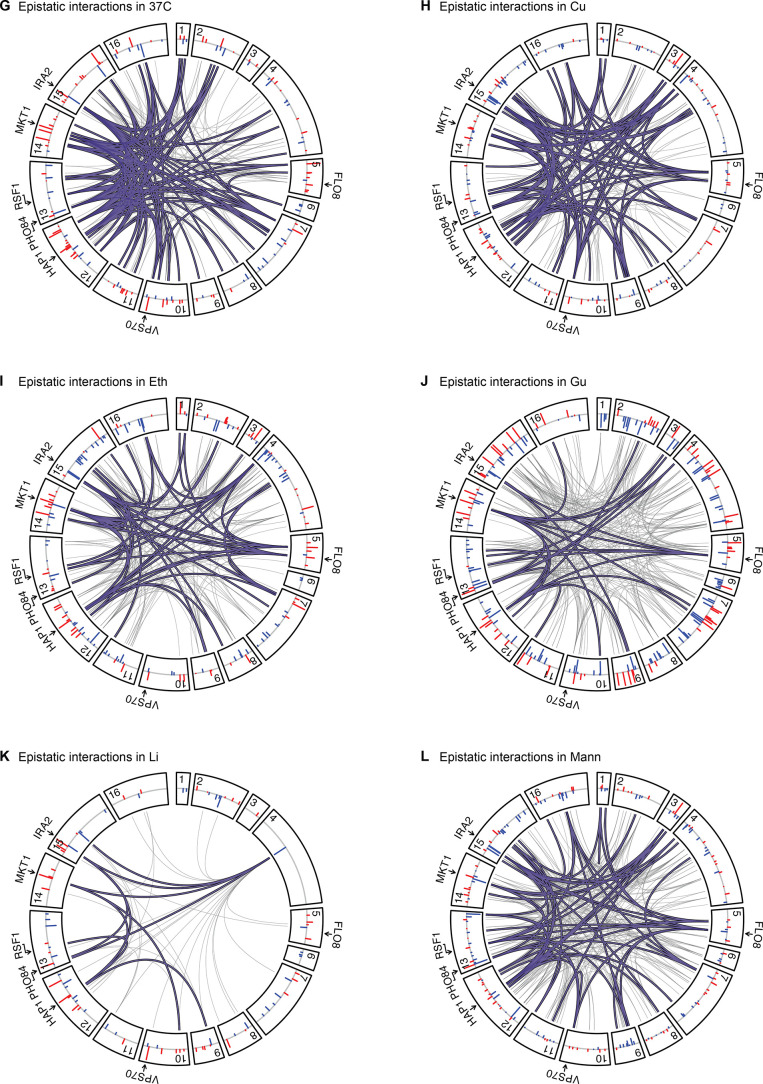
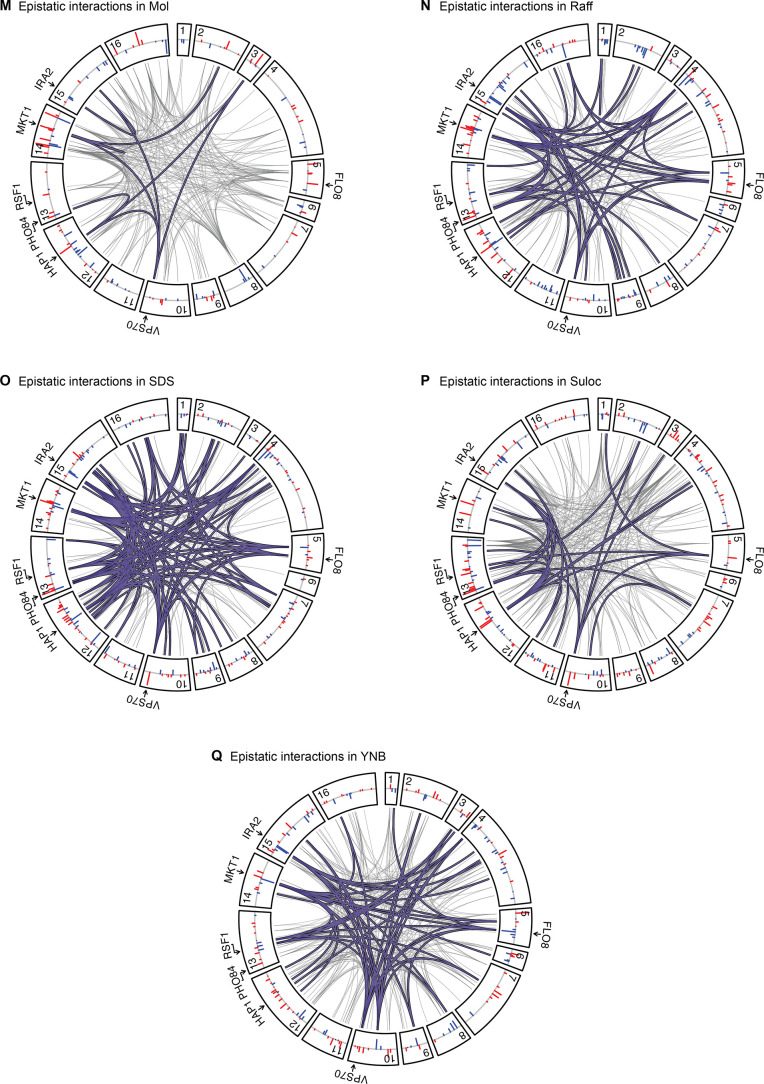
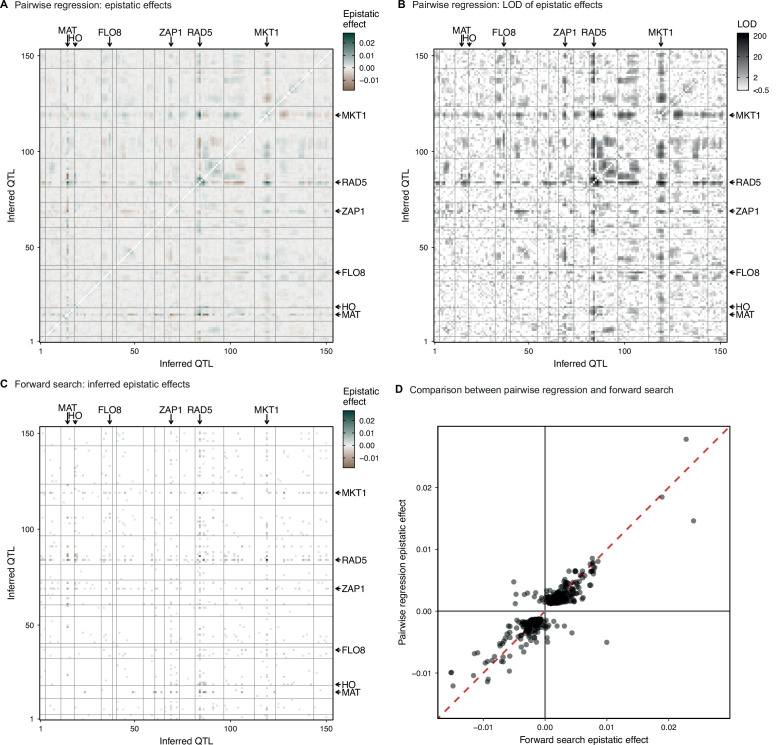
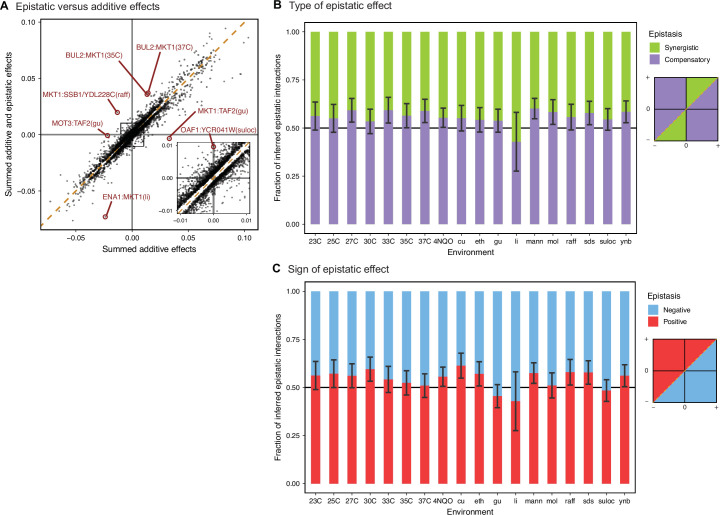
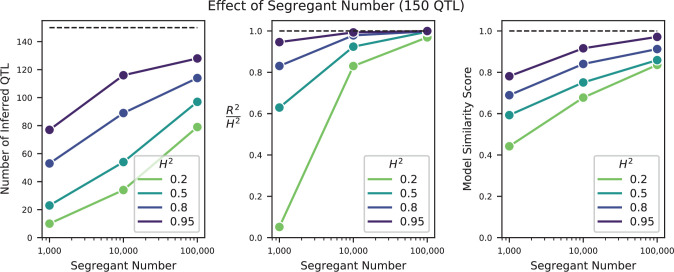
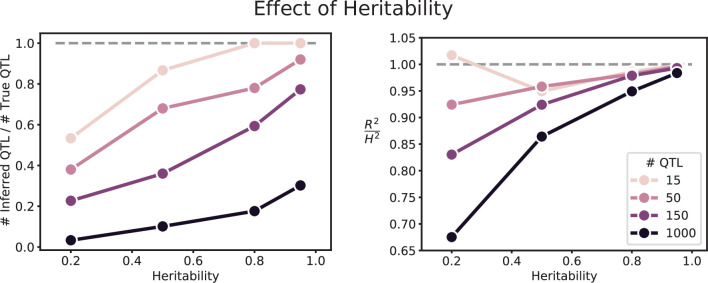
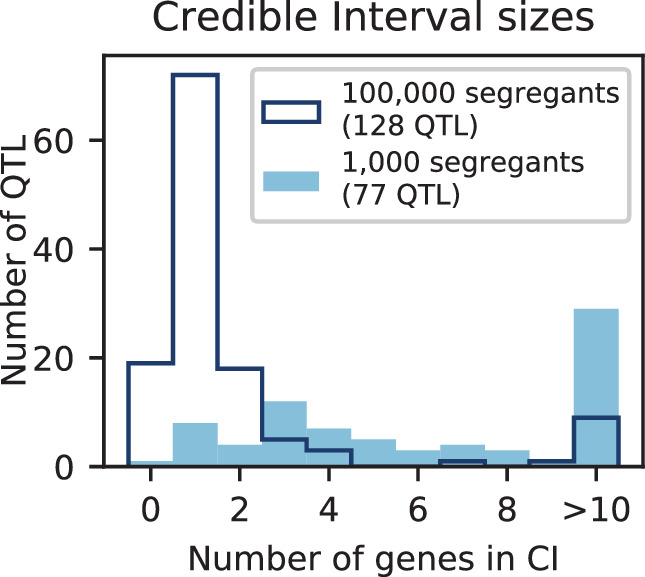
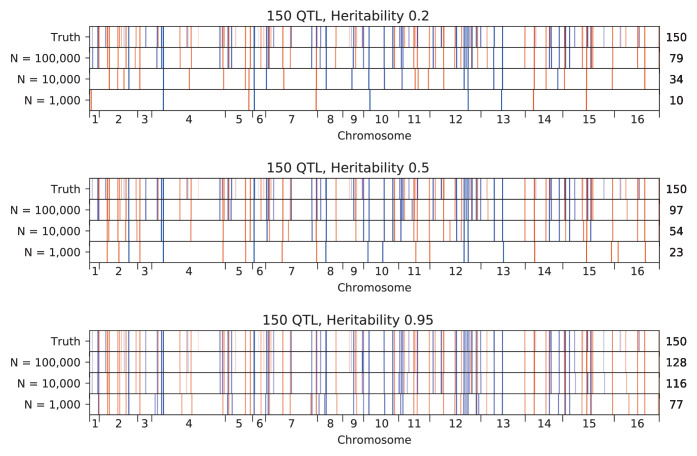
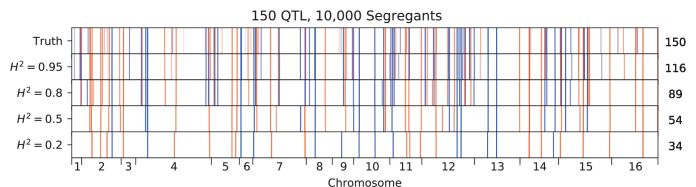
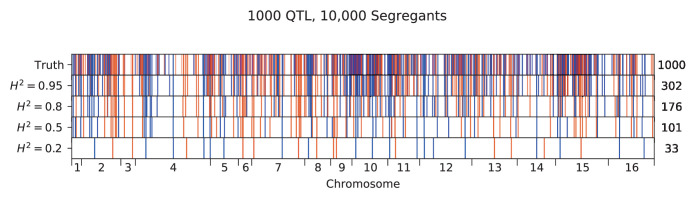
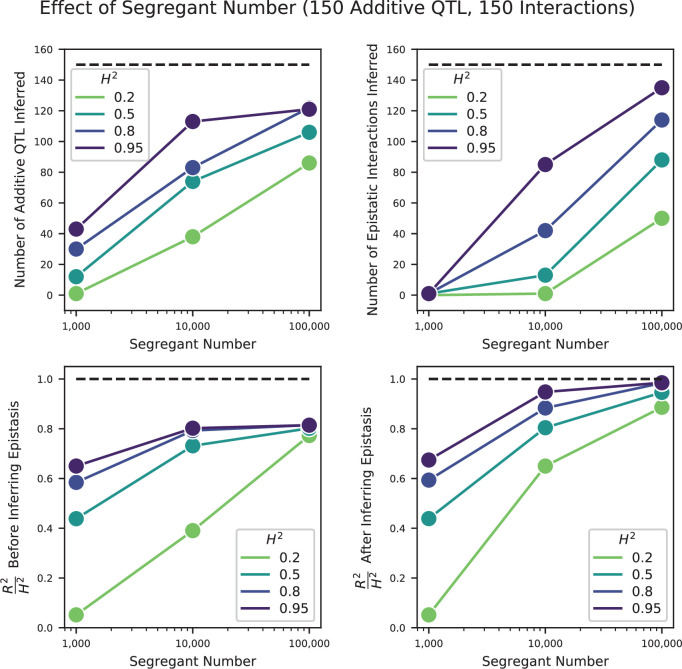
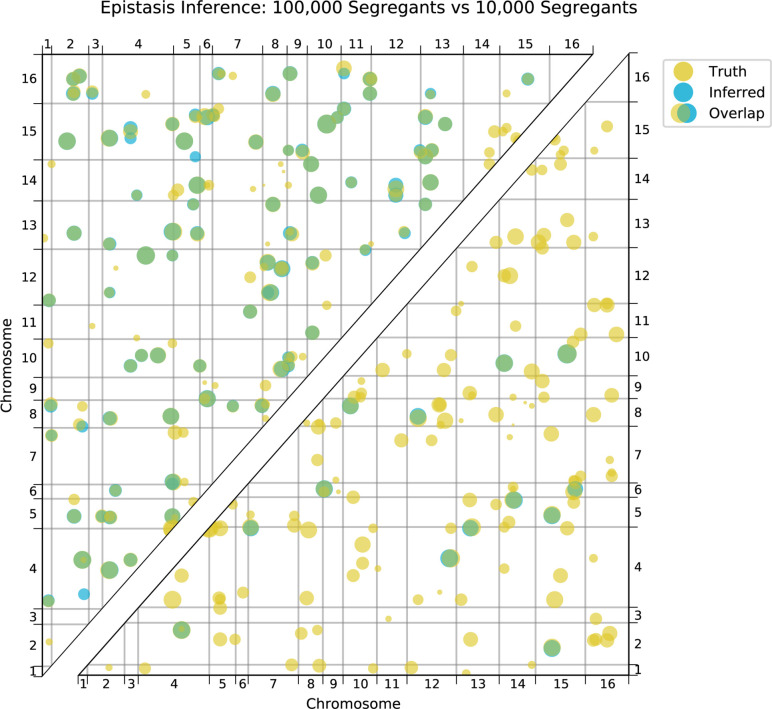
Mapping the genetic basis of complex traits is critical to uncovering the biological mechanisms that underlie disease and other phenotypes. Genome-wide association studies (GWAS) in humans and quantitative trait locus (QTL) mapping in model organisms can now explain much of the observed heritability in many traits, allowing us to predict phenotype from genotype. However, constraints on power due to statistical confounders in large GWAS and smaller sample sizes in QTL studies still limit our ability to resolve numerous small-effect variants, map them to causal genes, identify pleiotropic effects across multiple traits, and infer non-additive interactions between loci (epistasis). Here, we introduce barcoded bulk quantitative trait locus (BB-QTL) mapping, which allows us to construct, genotype, and phenotype 100,000 offspring of a budding yeast cross, two orders of magnitude larger than the previous state of the art. We use this panel to map the genetic basis of eighteen complex traits, finding that the genetic architecture of these traits involves hundreds of small-effect loci densely spaced throughout the genome, many with widespread pleiotropic effects across multiple traits. Epistasis plays a central role, with thousands of interactions that provide insight into genetic networks. By dramatically increasing sample size, BB-QTL mapping demonstrates the potential of natural variants in high-powered QTL studies to reveal the highly polygenic, pleiotropic, and epistatic architecture of complex traits.
Keywords: S. cerevisiae; epistasis; evolutionary biology; genetics; genomics; pleiotropy; polygenic traits; quantitative trait loci.
© 2022, Nguyen Ba et al.
Conflict of interest statement
AN, KL, AR, SG, DT, FM, MD No competing interests declared
Figures
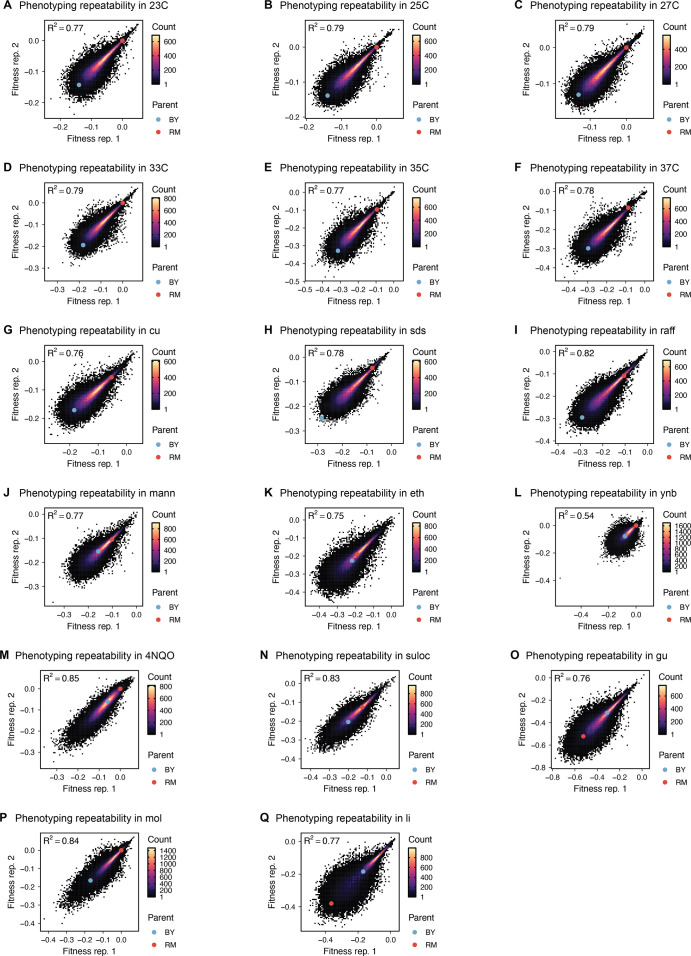
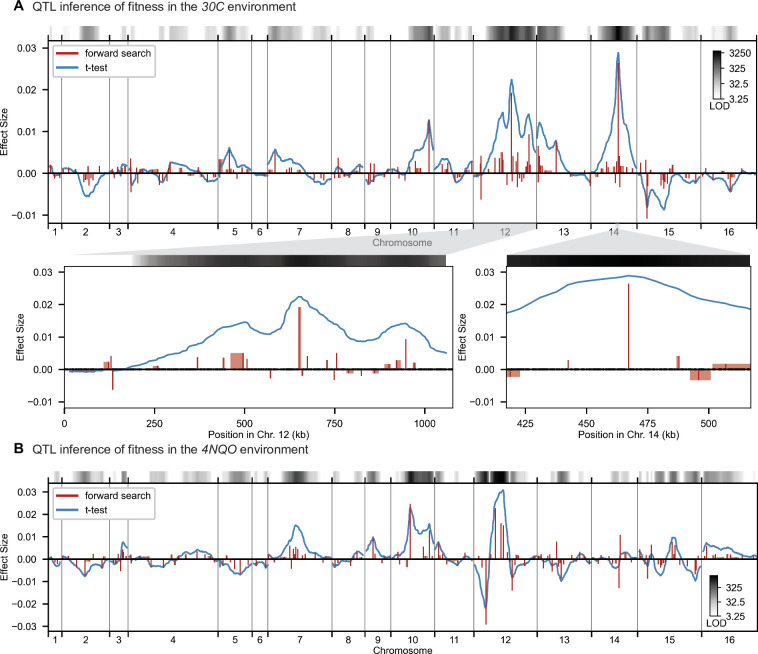
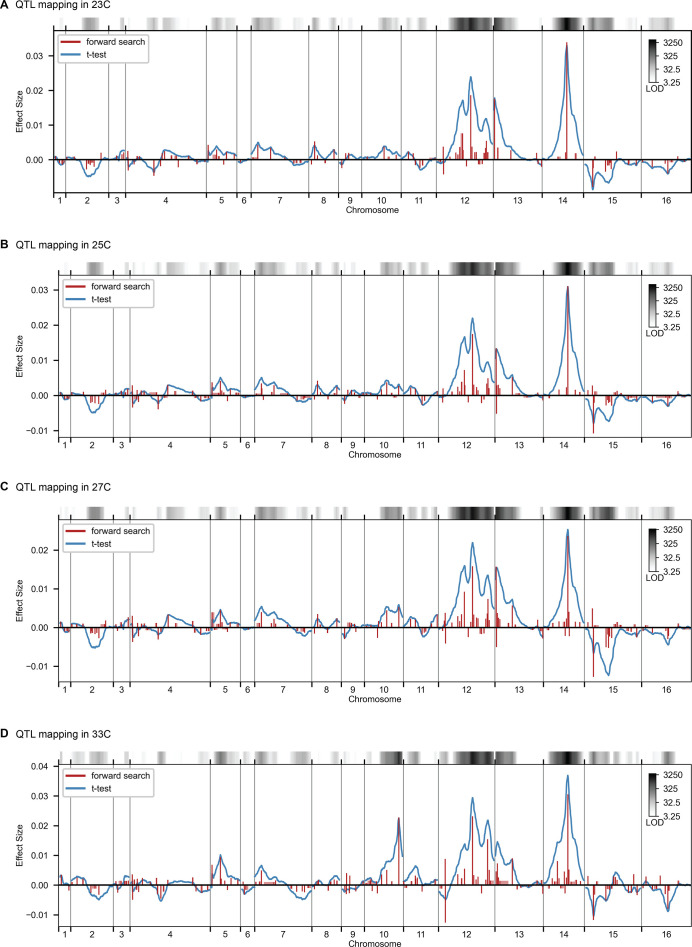
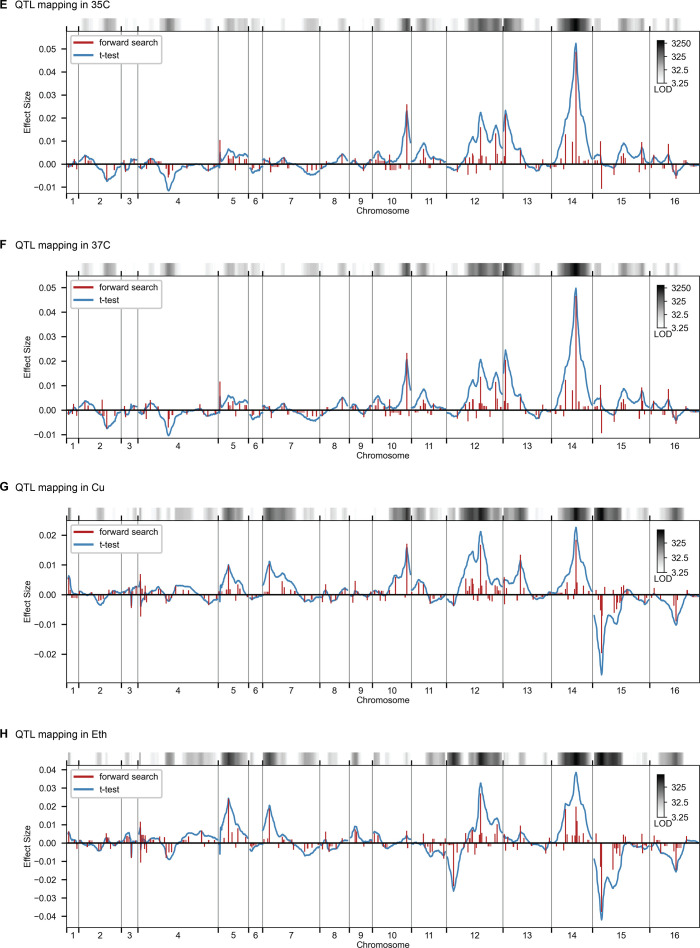
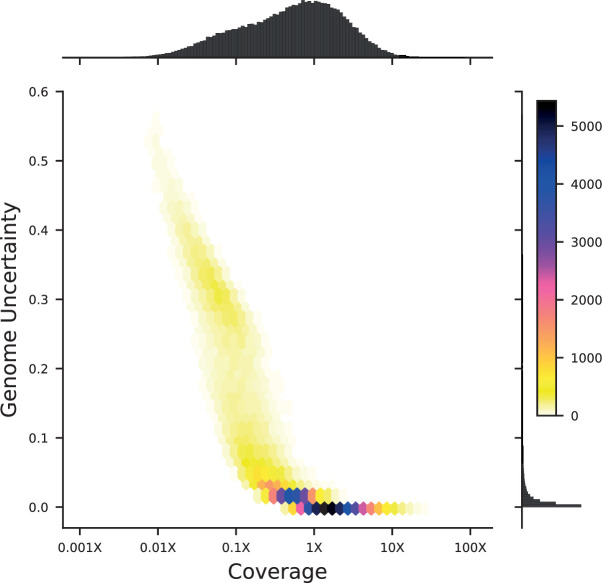
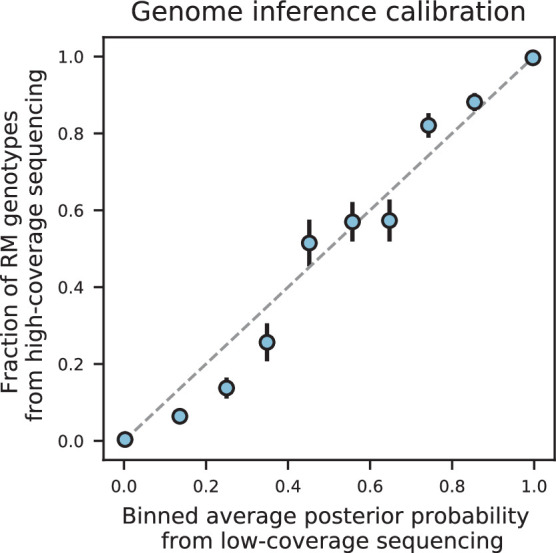
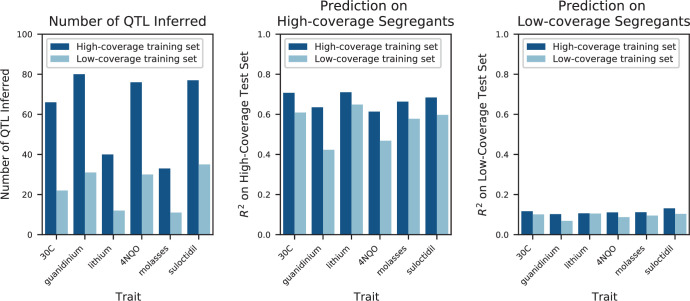
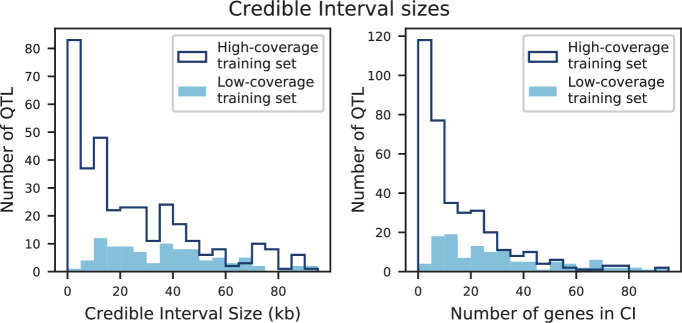
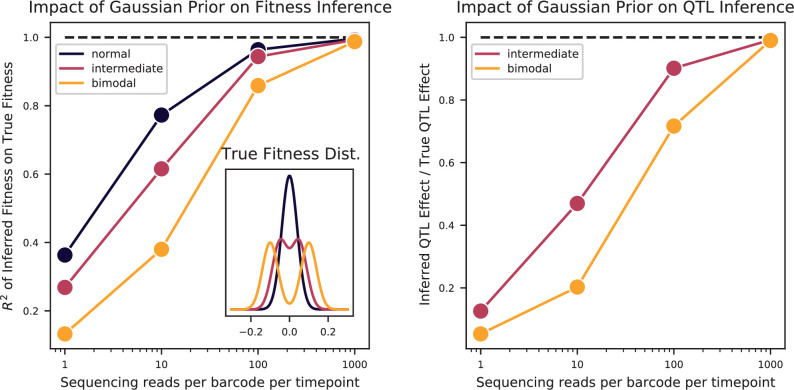
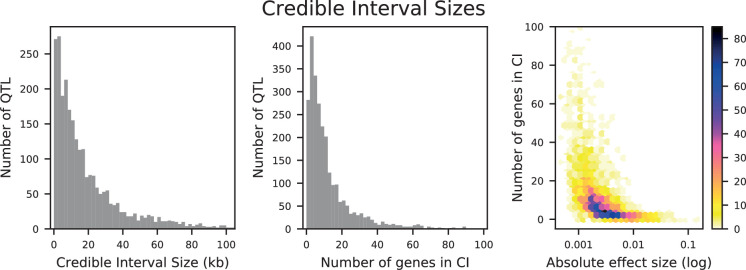
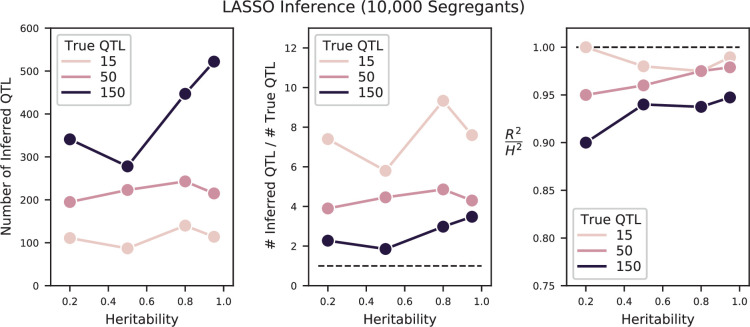
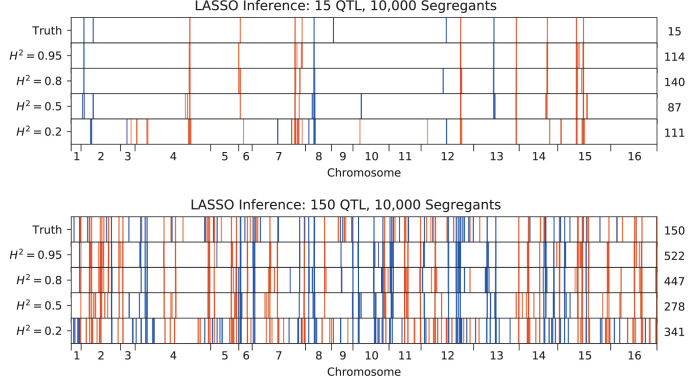
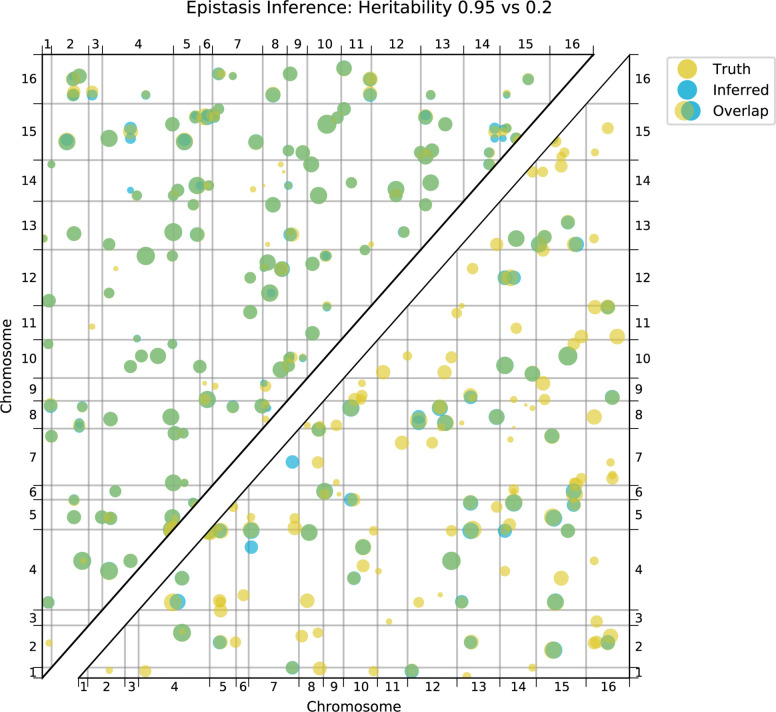
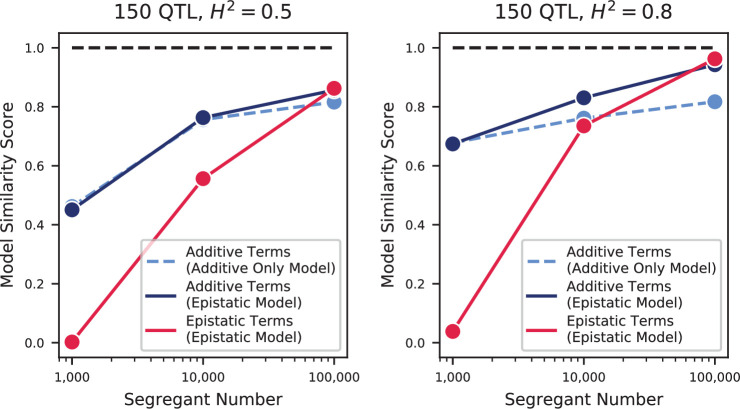
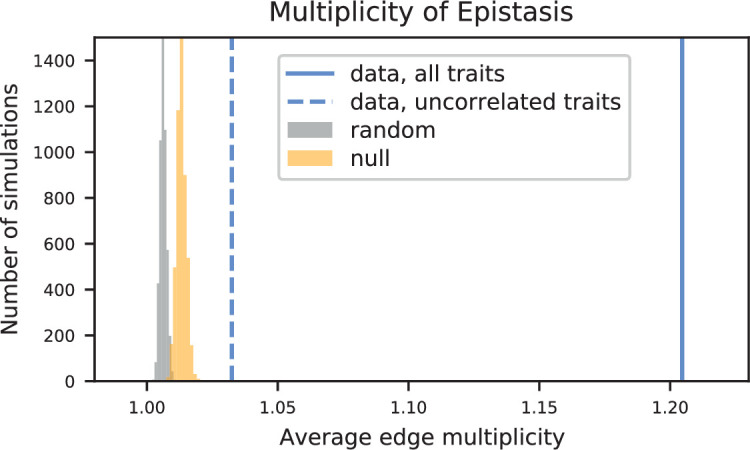
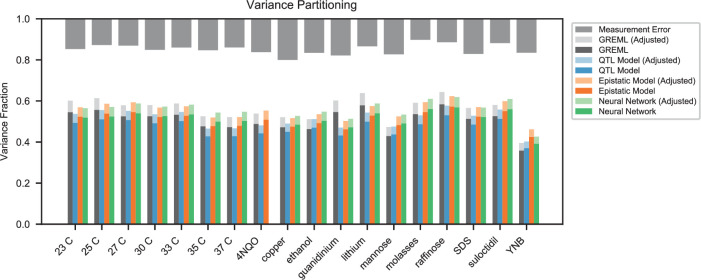
References
-
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, Ghemawat S, Goodfellow I, Harp A, Irving G, Isard M, Jia Y, Jozefowicz R, Kaiser L, Kudlur M, Levenberg J. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. 2015. [September 20, 2021]. https://www.tensorflow.org
-
- Adey A, Morrison HG, Asan XX, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, Shendure J. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biology. 2010;11:R119. doi: 10.1186/gb-2010-11-12-r119. - DOI - PMC - PubMed
-
- Allen DM. The Relationship Between Variable Selection and Data Agumentation and a Method for Prediction. Technometrics. 1974;16:125–127. doi: 10.1080/00401706.1974.10489157. - DOI
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
