

Enhancing Sampling of Water Rehydration on Ligand Binding: A Comparison of Techniques
- PMID: 35148093
- PMCID: PMC9241631
- DOI: 10.1021/acs.jctc.1c00590
Enhancing Sampling of Water Rehydration on Ligand Binding: A Comparison of Techniques
Abstract
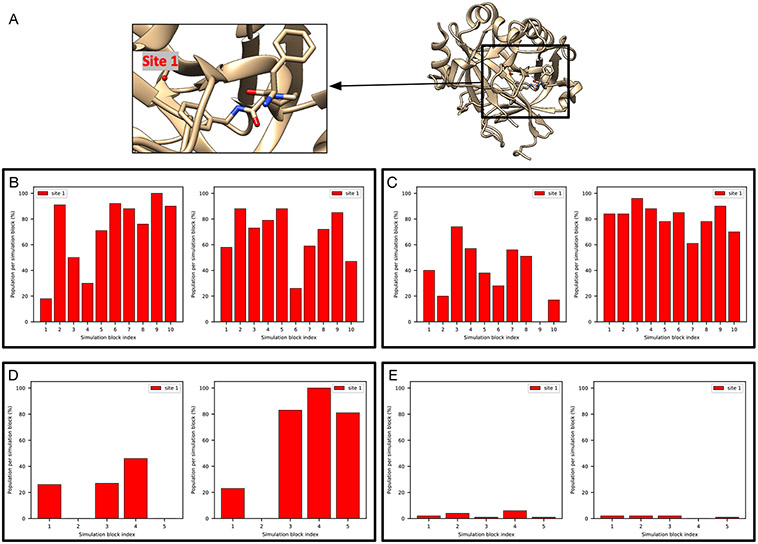
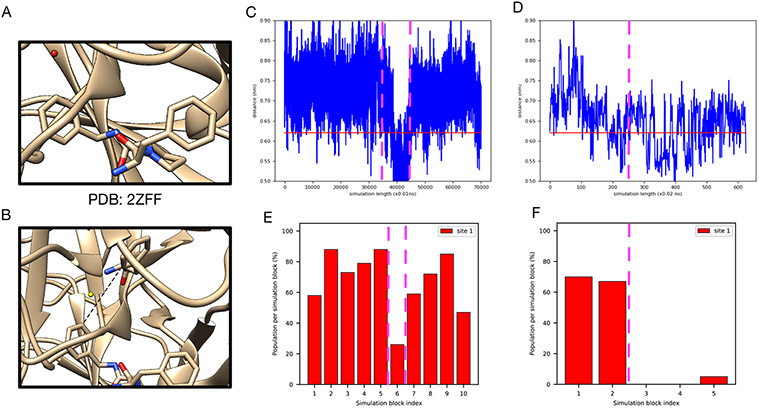
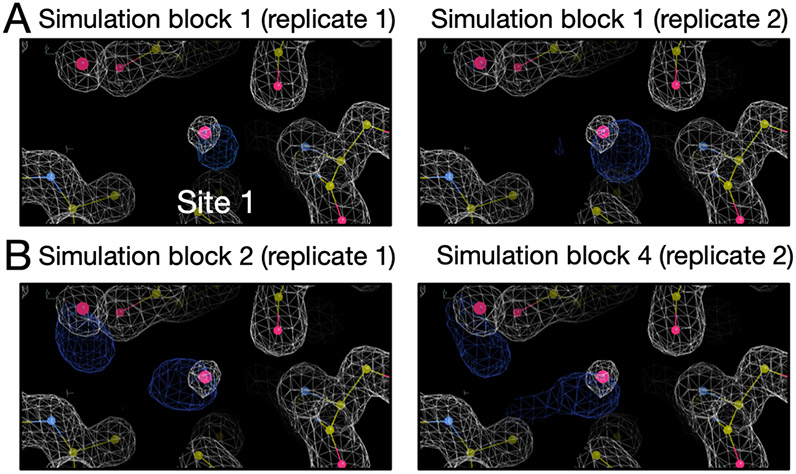
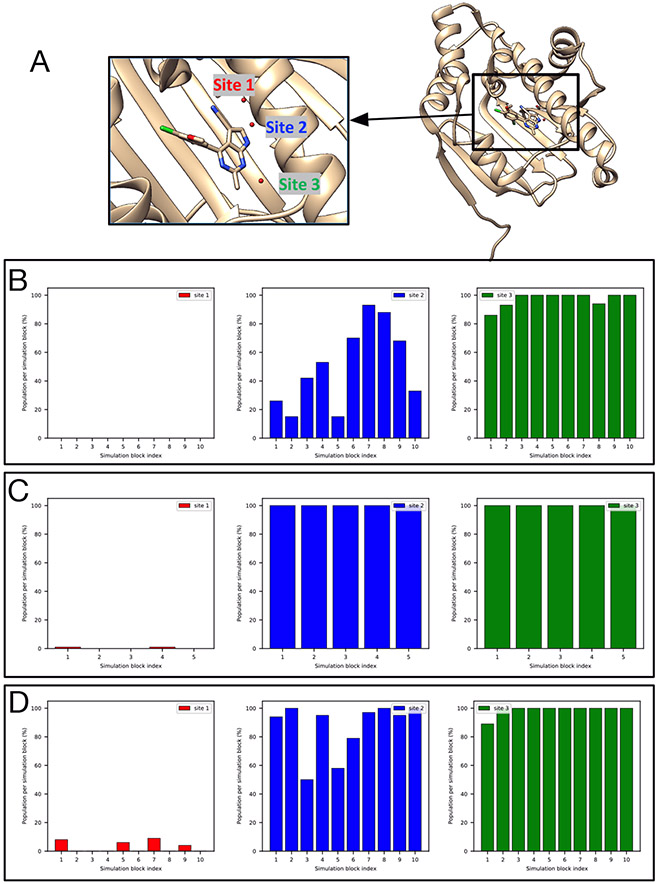
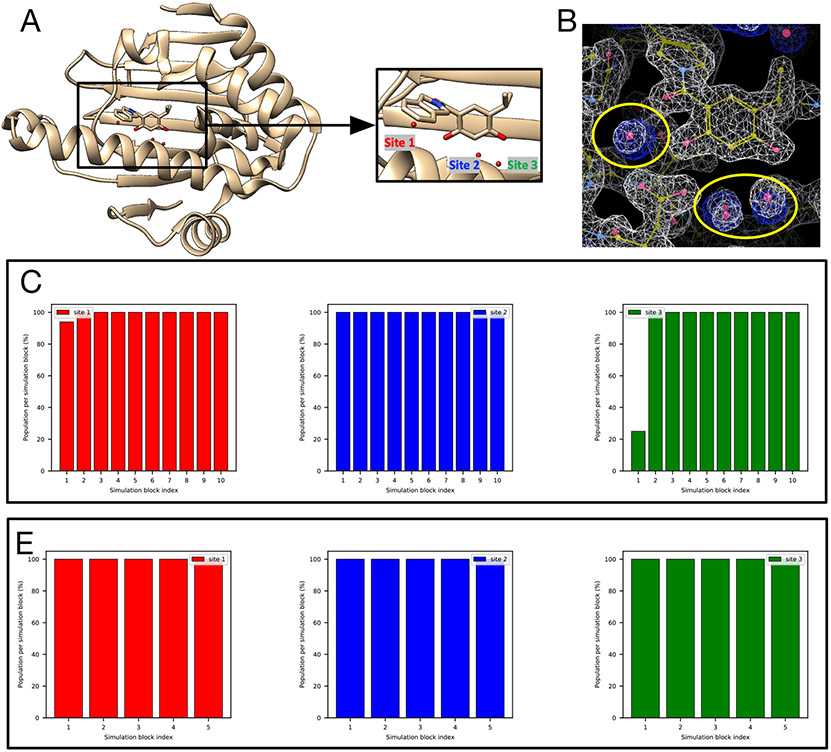
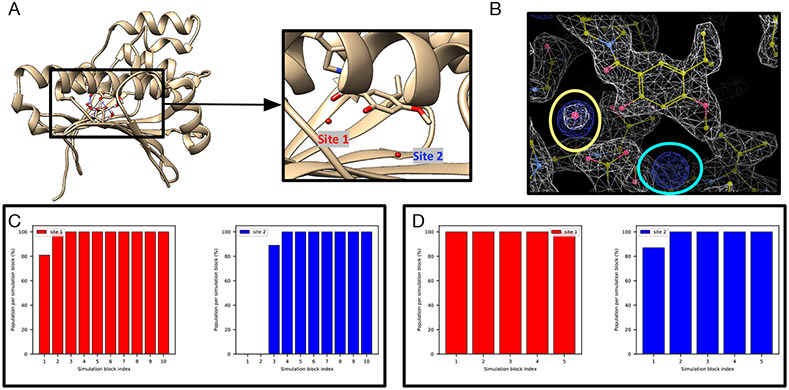
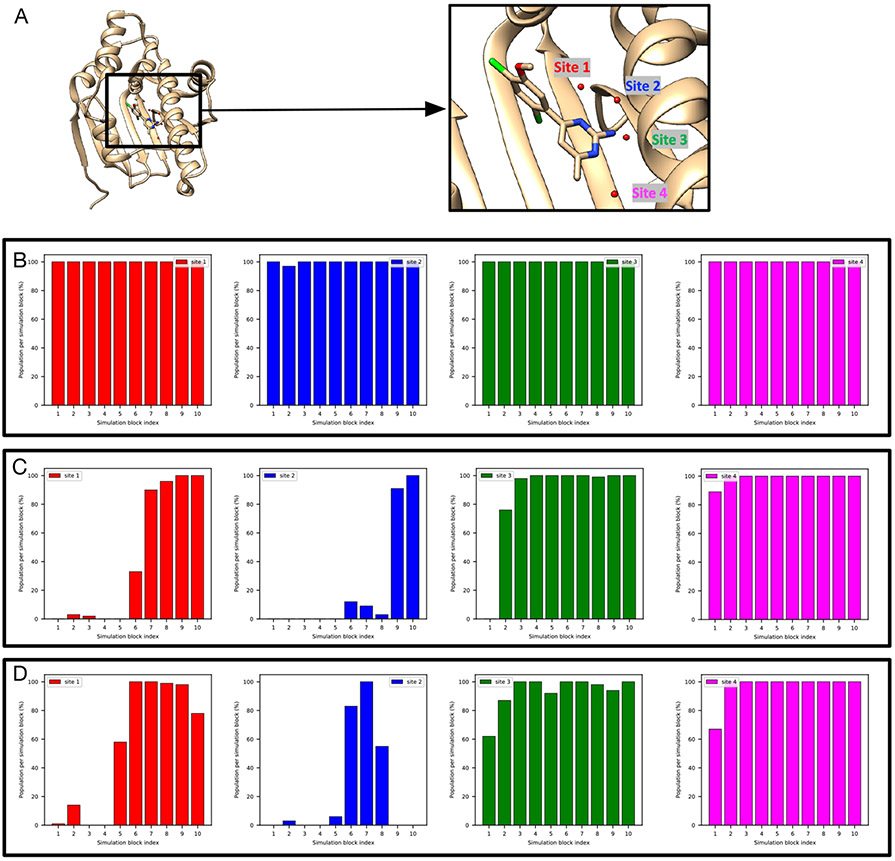
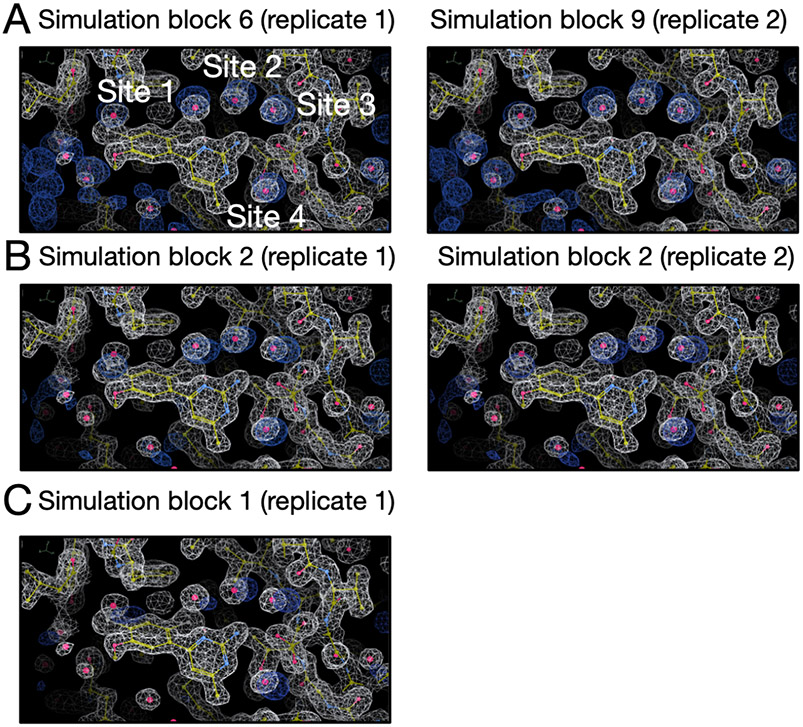
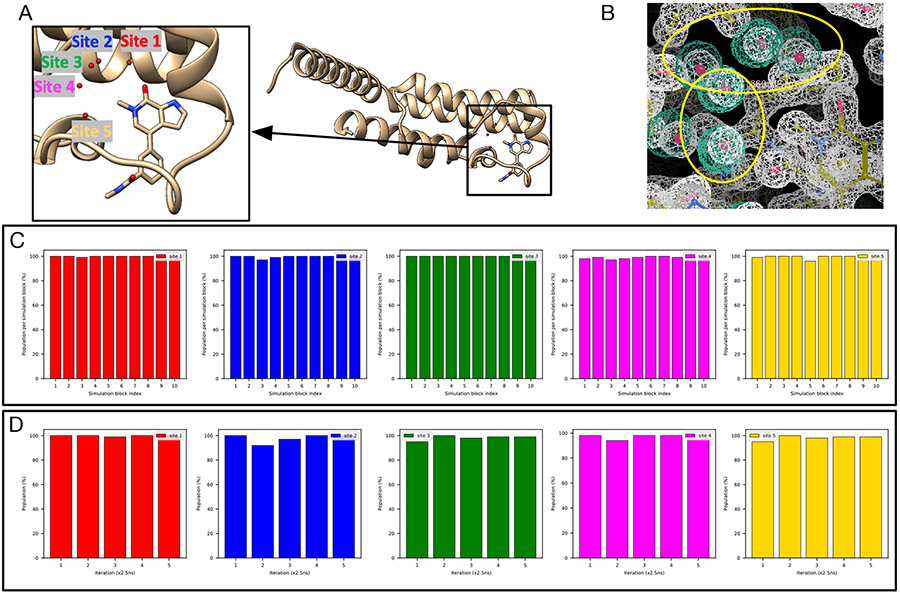
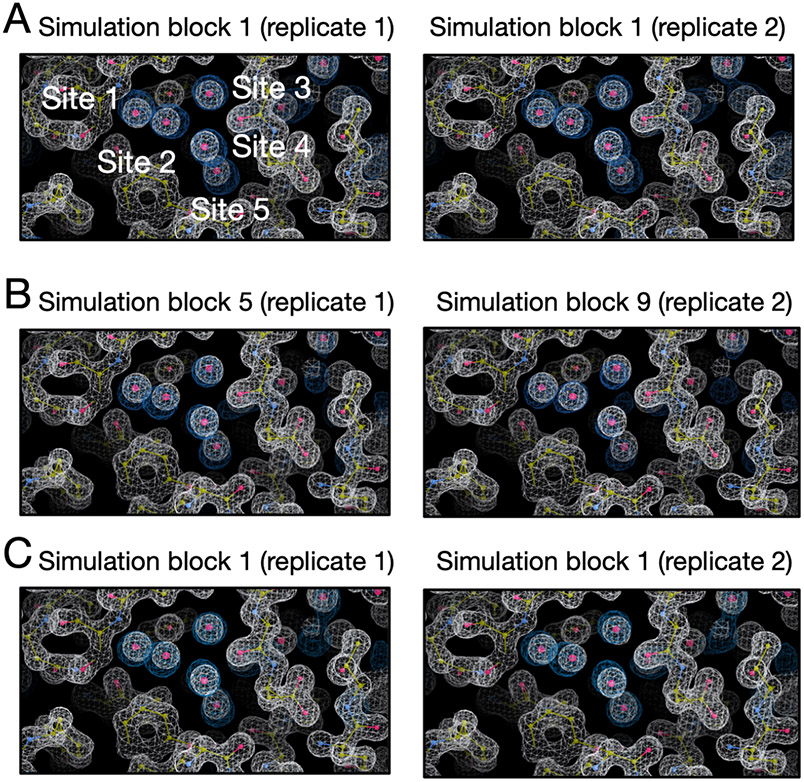
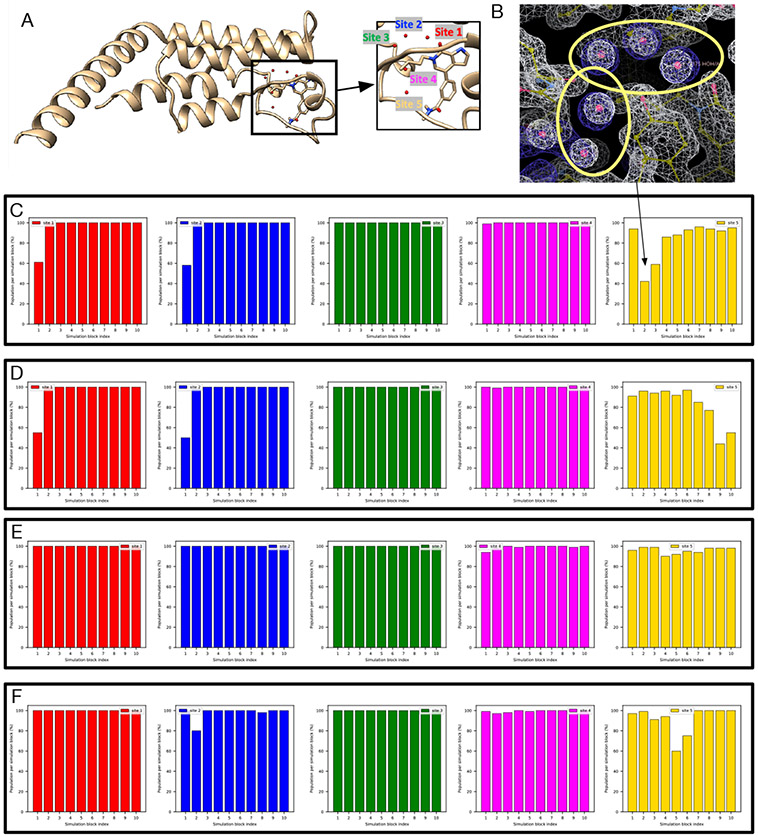
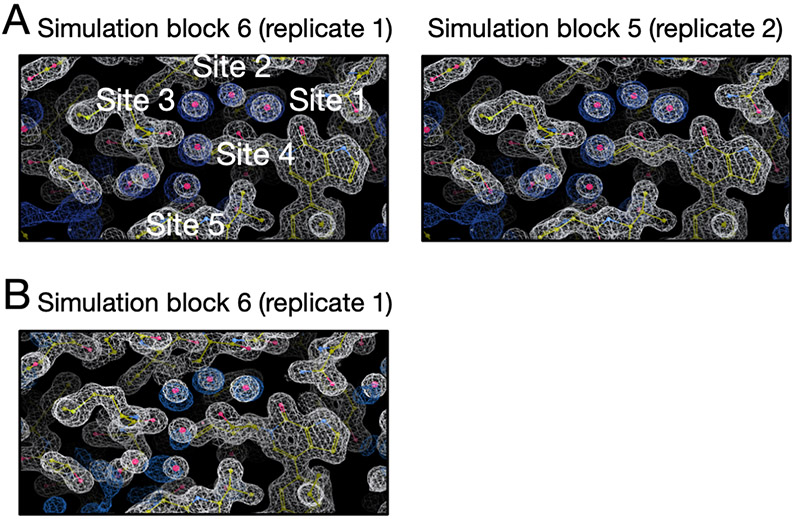
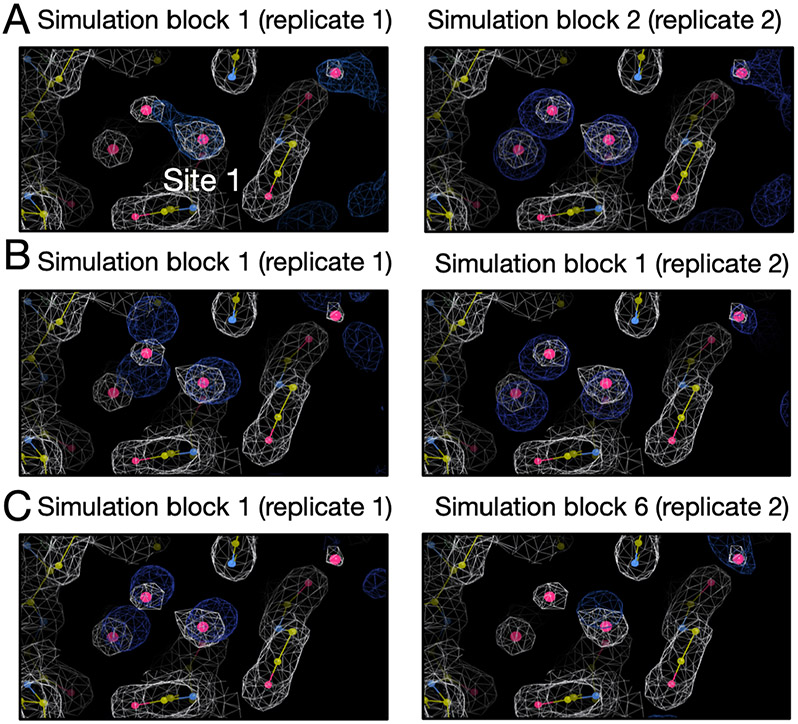
Water often plays a key role in protein structure, molecular recognition, and mediating protein-ligand interactions. Thus, free energy calculations must adequately sample water motions, which often proves challenging in typical MD simulation time scales. Thus, the accuracy of methods relying on MD simulations ends up limited by slow water sampling. Particularly, as a ligand is removed or modified, bulk water may not have time to fill or rearrange in the binding site. In this work, we focus on several molecular dynamics (MD) simulation-based methods attempting to help rehydrate buried water sites: BLUES, using nonequilibrium candidate Monte Carlo (NCMC); grand, using grand canonical Monte Carlo (GCMC); and normal MD. We assess the accuracy and efficiency of these methods in rehydrating target water sites. We selected a range of systems with varying numbers of waters in the binding site, as well as those where water occupancy is coupled to the identity or binding mode of the ligand. We analyzed the rehydration of buried water sites in binding pockets using both clustering of trajectories and direct analysis of electron density maps. Our results suggest both BLUES and grand enhance water sampling relative to normal MD and grand is more robust than BLUES, but also that water sampling remains a major challenge for all of the methods tested. The lessons we learned for these methods and systems are discussed.
Figures
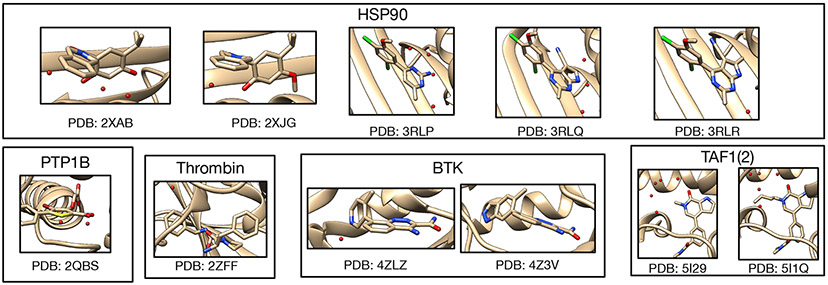
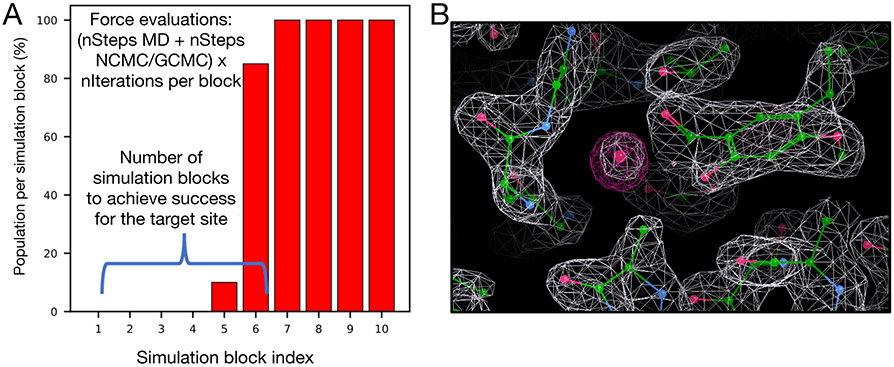
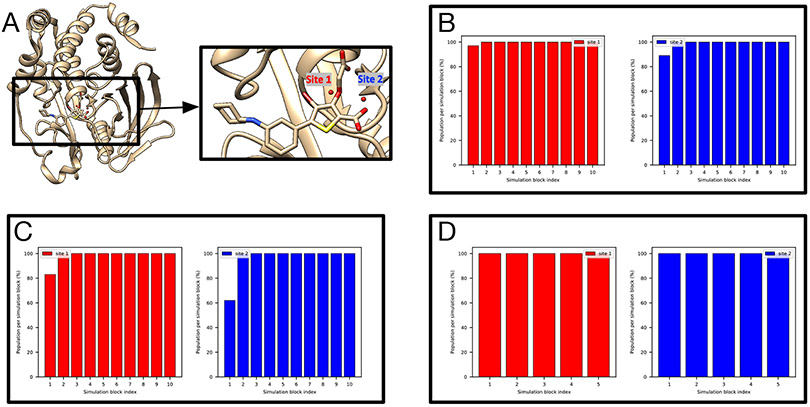
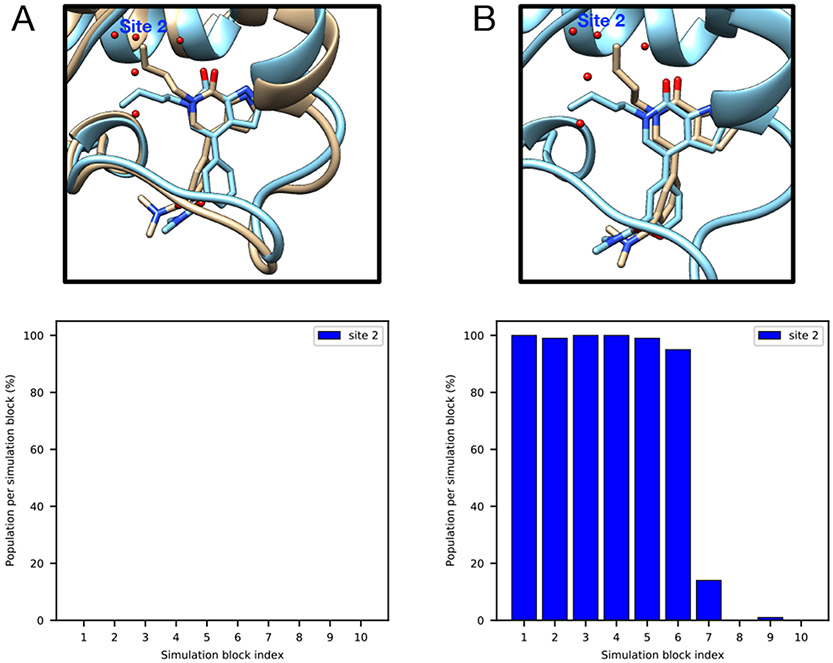
Similar articles
-
Enhancing sampling of water rehydration upon ligand binding using variants of grand canonical Monte Carlo.J Comput Aided Mol Des. 2022 Oct;36(10):767-779. doi: 10.1007/s10822-022-00479-w. Epub 2022 Oct 6. J Comput Aided Mol Des. 2022. PMID: 36198874 Free PMC article.
-
Enhancing water sampling of buried binding sites using nonequilibrium candidate Monte Carlo.J Comput Aided Mol Des. 2021 Feb;35(2):167-177. doi: 10.1007/s10822-020-00344-8. Epub 2020 Sep 24. J Comput Aided Mol Des. 2021. PMID: 32968887 Free PMC article.
-
Binding Modes of Ligands Using Enhanced Sampling (BLUES): Rapid Decorrelation of Ligand Binding Modes via Nonequilibrium Candidate Monte Carlo.J Phys Chem B. 2018 May 31;122(21):5579-5598. doi: 10.1021/acs.jpcb.7b11820. Epub 2018 Mar 12. J Phys Chem B. 2018. PMID: 29486559 Free PMC article.
-
Current Tools and Methods in Molecular Dynamics (MD) Simulations for Drug Design.Curr Med Chem. 2016;23(34):3909-3924. doi: 10.2174/0929867323666160530144742. Curr Med Chem. 2016. PMID: 27237821 Review.
-
Taming Rugged Free Energy Landscapes Using an Average Force.Acc Chem Res. 2019 Nov 19;52(11):3254-3264. doi: 10.1021/acs.accounts.9b00473. Epub 2019 Nov 4. Acc Chem Res. 2019. PMID: 31680510 Review.
Cited by
-
An open-source molecular builder and free energy preparation workflow.Commun Chem. 2022;5(1):136. doi: 10.1038/s42004-022-00754-9. Epub 2022 Oct 27. Commun Chem. 2022. PMID: 36320862 Free PMC article.
-
Conformational Selectivity of ITK Inhibitors: Insights from Molecular Dynamics Simulations.J Chem Inf Model. 2023 Dec 25;63(24):7860-7872. doi: 10.1021/acs.jcim.3c01352. Epub 2023 Dec 9. J Chem Inf Model. 2023. PMID: 38069816 Free PMC article.
-
Molecular-dynamics simulation methods for macromolecular crystallography.Acta Crystallogr D Struct Biol. 2023 Jan 1;79(Pt 1):50-65. doi: 10.1107/S2059798322011871. Epub 2023 Jan 1. Acta Crystallogr D Struct Biol. 2023. PMID: 36601807 Free PMC article.
-
Relative Binding Free Energy between Chemically Distant Compounds Using a Bidirectional Nonequilibrium Approach.J Chem Theory Comput. 2022 Jun 14;18(6):4014-4026. doi: 10.1021/acs.jctc.2c00295. Epub 2022 Jun 1. J Chem Theory Comput. 2022. PMID: 35642423 Free PMC article.
-
Databases of ligand-binding pockets and protein-ligand interactions.Comput Struct Biotechnol J. 2024 Mar 24;23:1320-1338. doi: 10.1016/j.csbj.2024.03.015. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 38585646 Free PMC article. Review.
References
-
- Ball P Water as an Active Constituent in Cell Biology. Chem. Rev 2008, 108, 74–108. - PubMed
-
- Levy Y; Onuchic JN Water Mediation in Portein Folding and Molecular Recognition. Annu. Rev. Biophys. Biomol. Struct 2006, 35, 389–415. - PubMed
-
- Ernst JA; Clubb RT; Zhou H-X; Gronenborn AM; Clore GM Demonstration of positionally disordered water within a protein hydrophobic cavity by NMR. Science 1995, 267, 1813–1817. - PubMed
-
- Nittinger E; Schneider N; Lange G; Rarey M Evidence of Water Molecules—A Statistical Evaluation of Water Molecules Based on Electron Density. J. Chem. Inf. Model 2015, 55, 771–783. - PubMed